amplicon-nf: Viewing outputs of the pipeline in EPI2ME
Viewing outputs of the amplicon-nf pipeline in EPI2ME
| Document: | ARTIC-amplicon_nf-outputs-guide-v1.0.0 |
| Creation Date: | 2025-08-27 |
| Author: | Sam Wilkinson |
| Licence: | Creative Commons Attribution 4.0 International License |
This guide is specifically for readers who have run already run amplicon-nf using EPI2ME and wish to view their output files inside of EPI2ME, if you have run the pipeline via the command line you may wish to check the documentation in the github repository for information about the outputs of the pipeline.
This guide also assumes that you left the “Outdir” parameter as the default value, if you changed this EPI2ME will not track your outputs, the files will be present but you will have to navigate to this directory with your file browser.
Once a pipeline completes successfully your client should look like this:
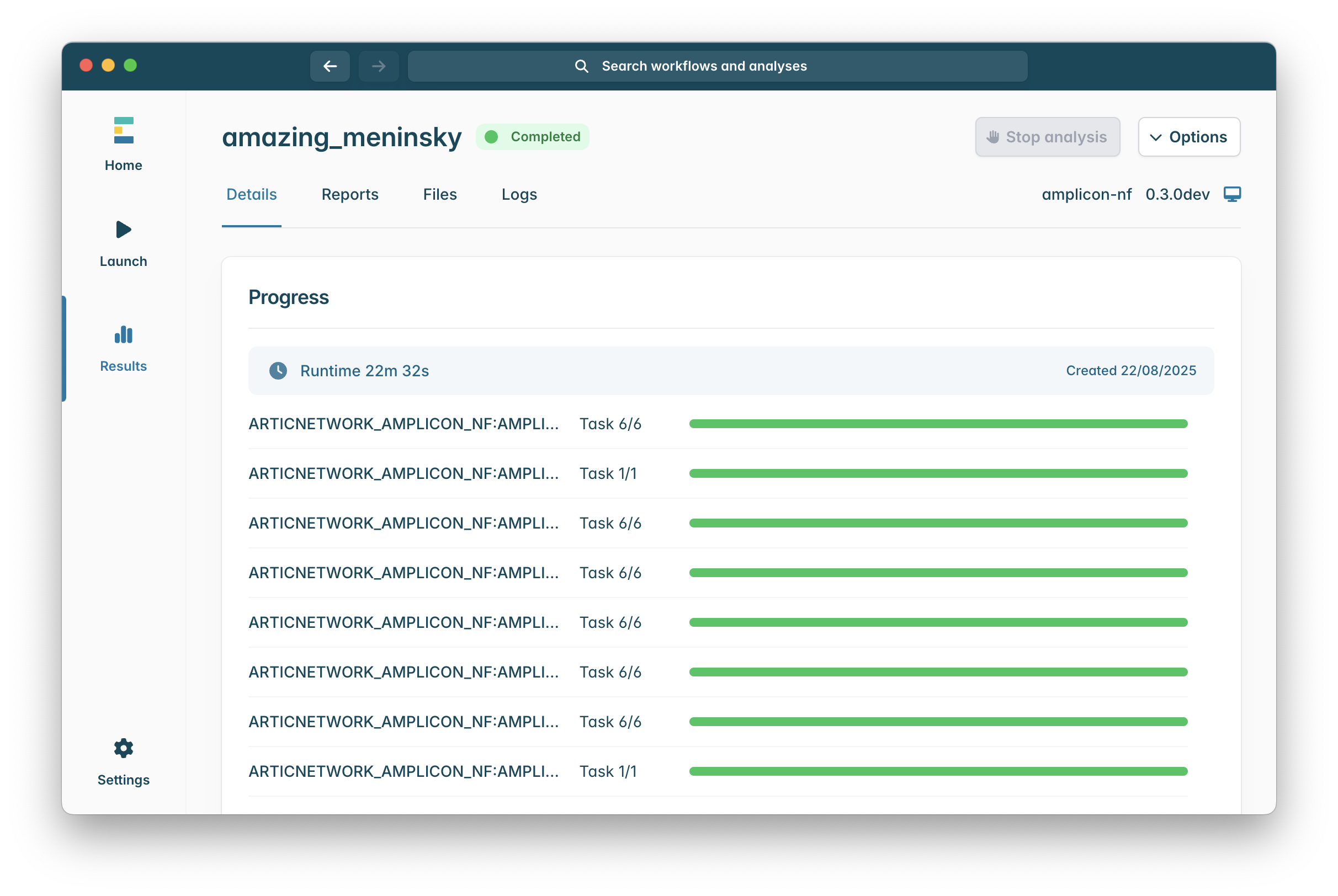
The important tabs for our purposes are “Reports” and “Files”.
Reports
To switch between reports, press the dropdown menu at the top of the “Reports” tab, this will show you a list of all available report files (EPI2ME searches the run directory for any HTML files).
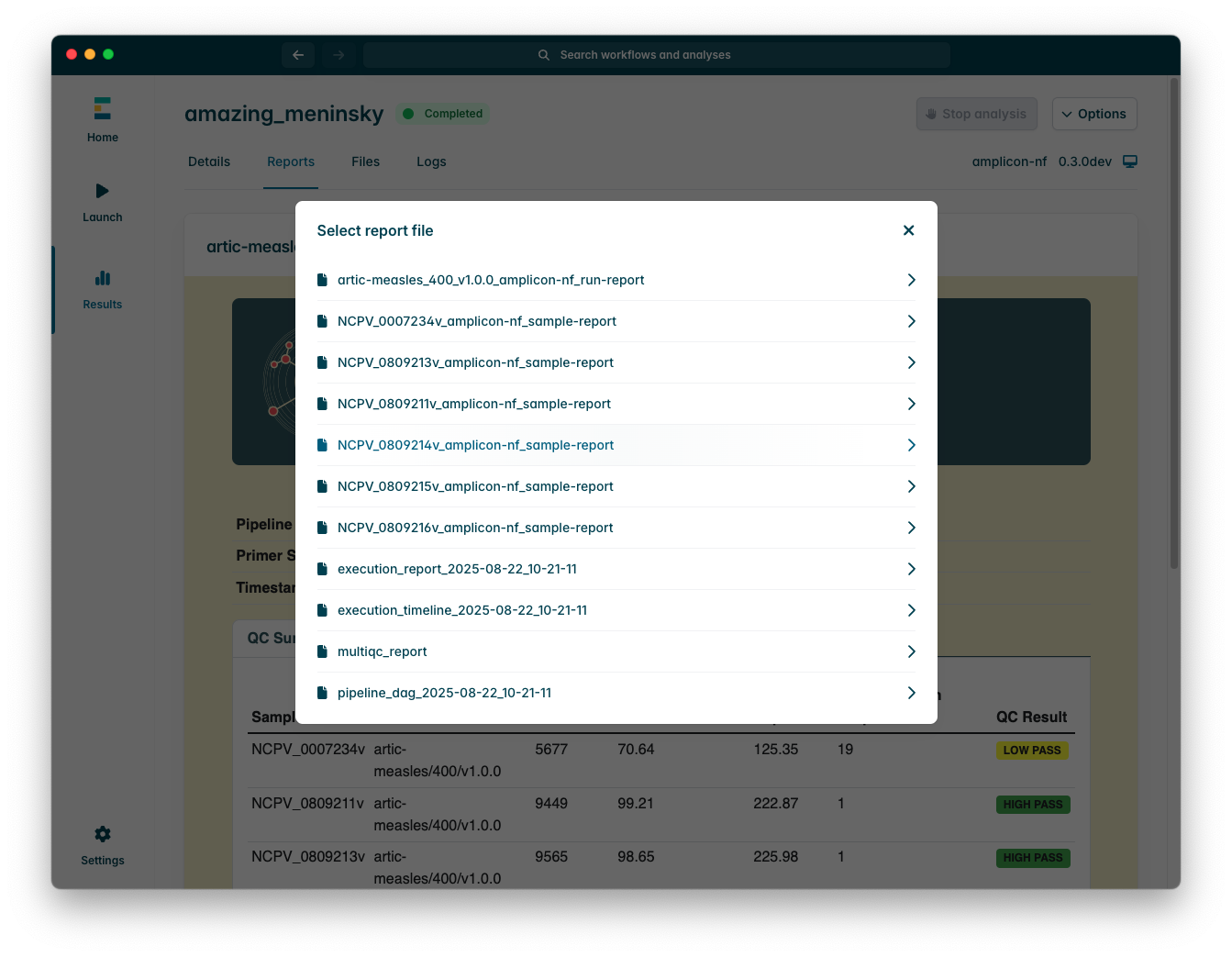
Report files are HTML documents which are useful for performing basic quality control (QC) of your sequencing run, there are two types produced by amplicon-nf;
- Run reports - These reports are designed to give you an overview of all of your samples at once.
- Sample reports - These reports give your information about a specific sample.
Run / Scheme Reports
If you are only using a single scheme in your run (the pipeline supports multi scheme runs) then you will only have a single run report since they are generated for each scheme provided within the samplesheet.
When you open a run report it should look like this:
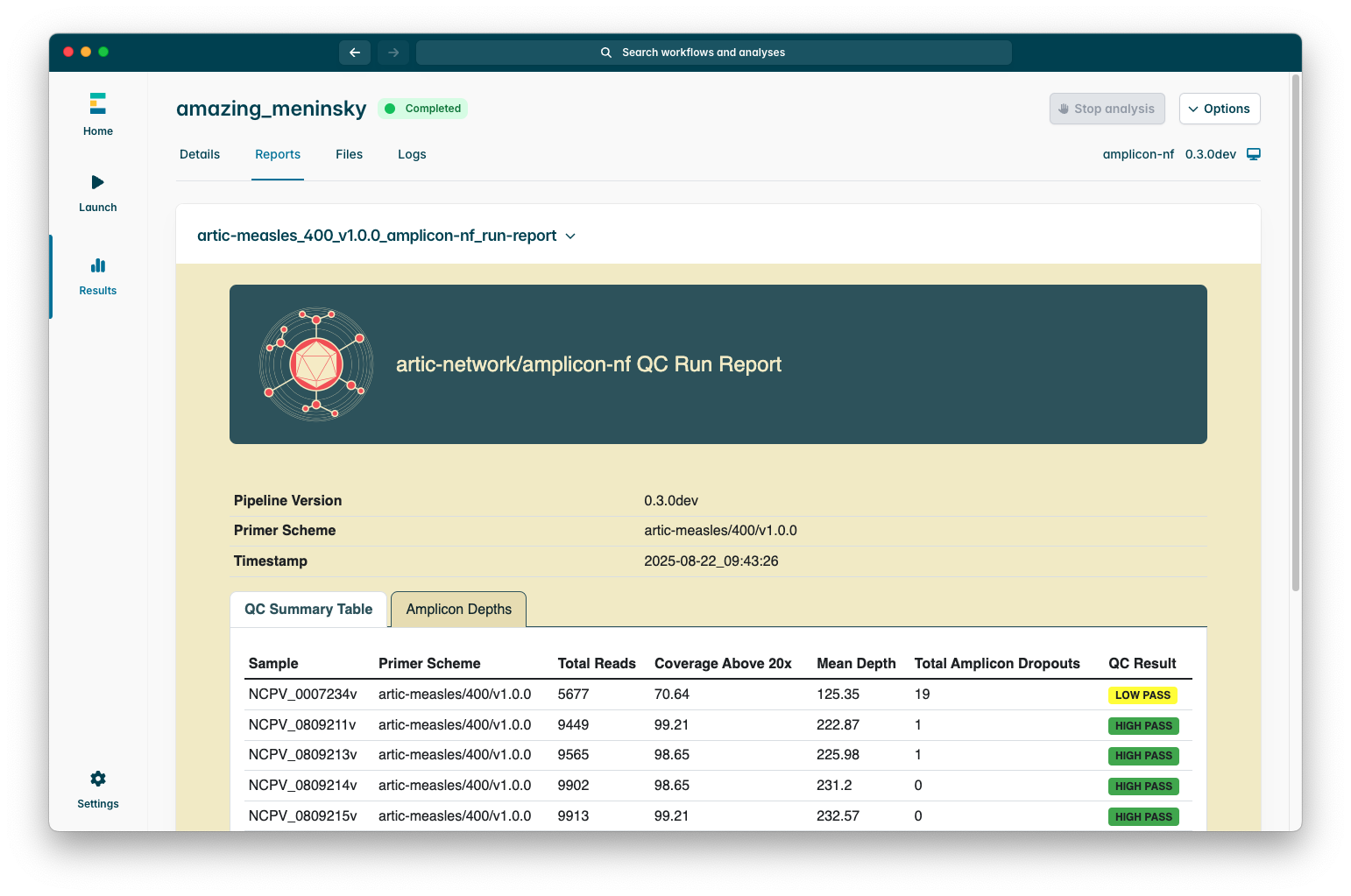
The table in the “QC Summary Table” quickly summarises how well each sample within the sequencing run performed, including total reads, the proportion of the genome which was covered at a depth above the “Min Coverage Depth” parameter (defaulting to 20), a count of amplicon dropouts, and a broad “QC Result” column which indicates whether the genome is a “Fail”, “Low Pass”, or “High Pass”.
The coverage thresholds for a sample to be considered a QC pass / fail are configurable using the “QC pass min coverage” and “QC pass high coverage” parameters.
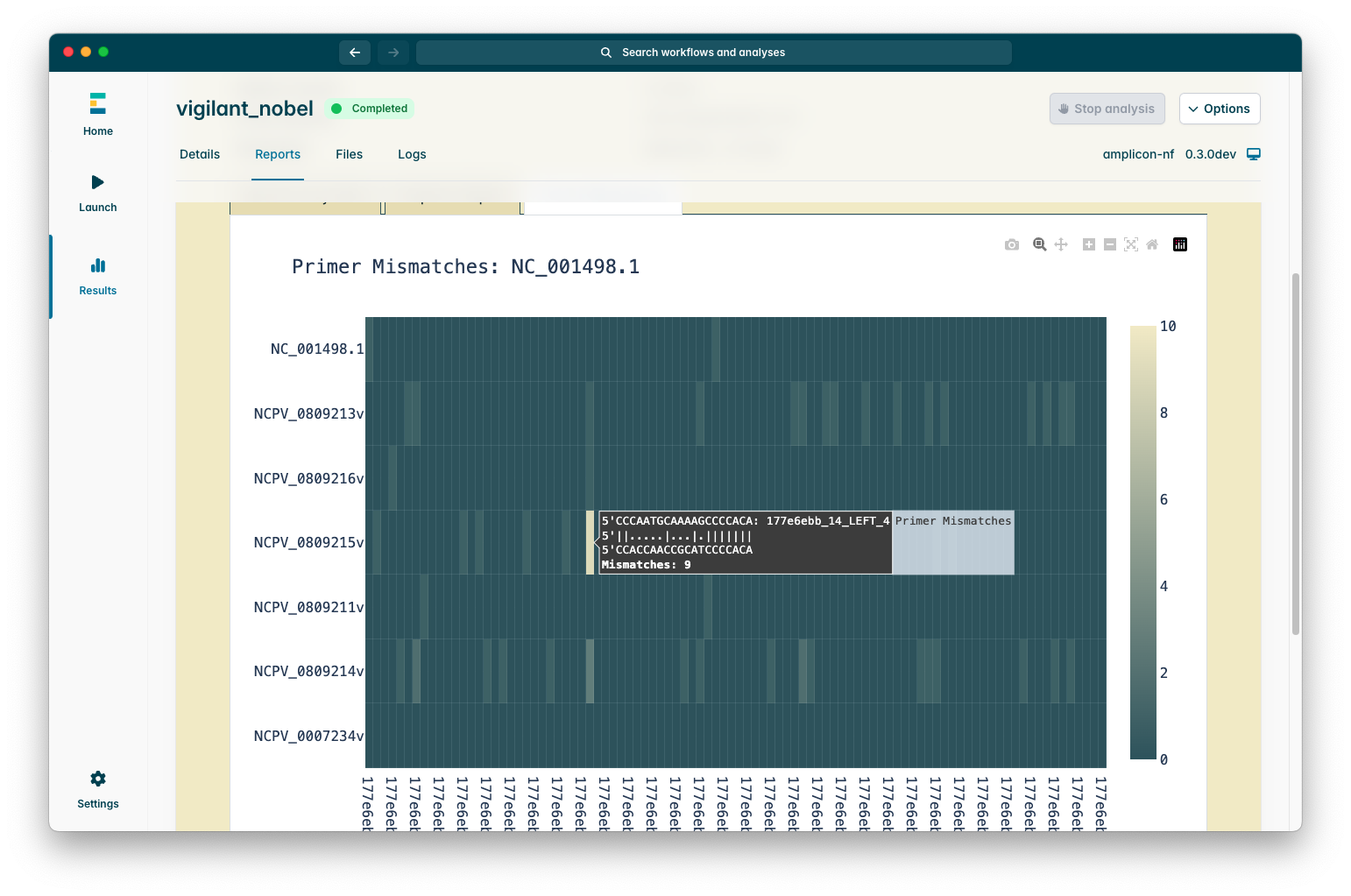
There is also an “Amplicon Depths Plot” tab which summarises how well each amplicon in each sample across the run performed, the colour scale is capped at the level of the “Min Coverage Depth” parameter, this is because this plot is designed to help identify amplicon dropouts (or patterns of amplicon dropouts) across a whole run at a glance.
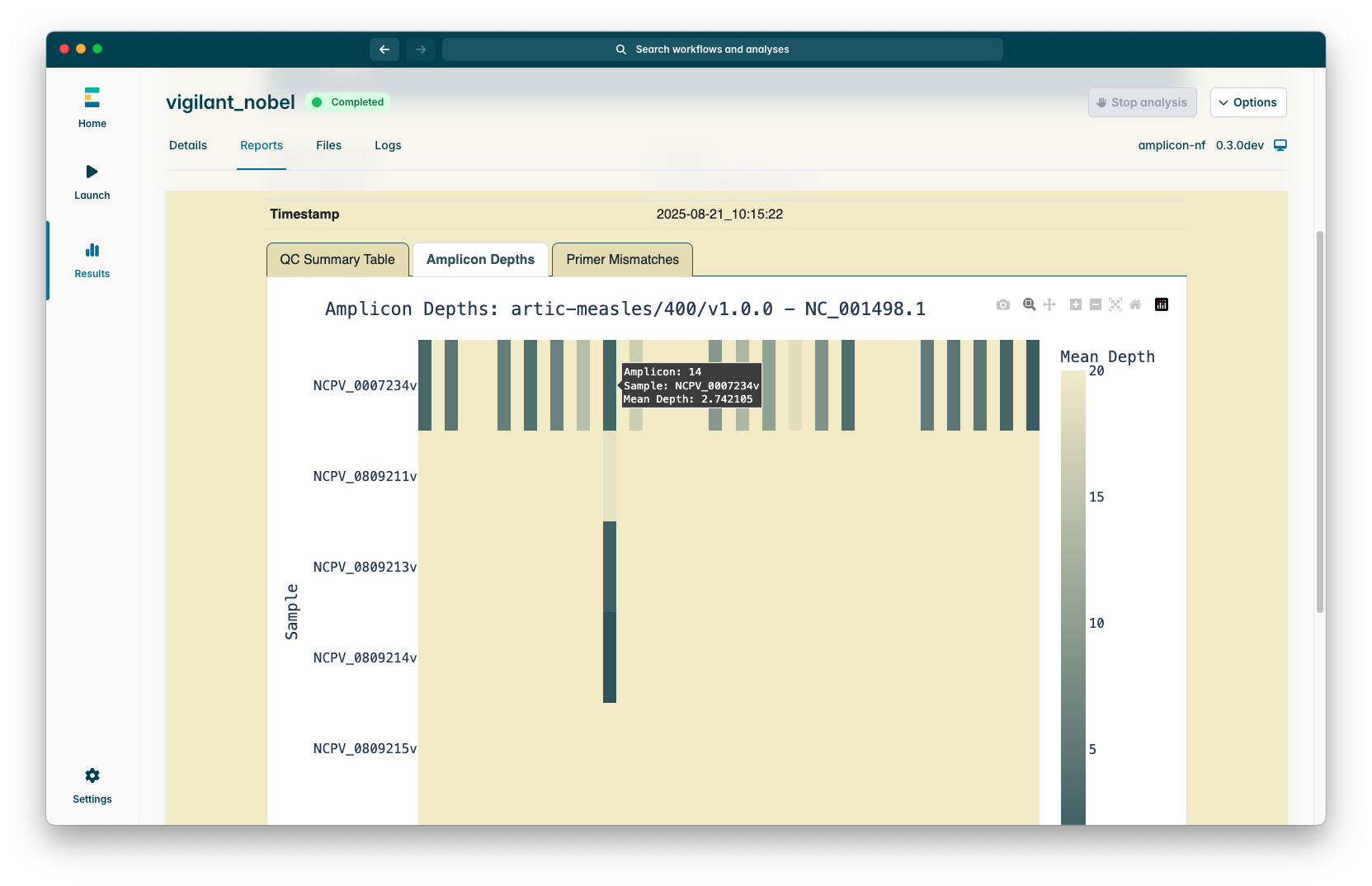
If you selected the “Primer mismatch plot” checkbox when launching the pipeline you will also see a “Primer mismatches” plot which attempts to detect positions where primer sites do not match the sequence and may lead to amplicon dropouts, this is not perfect due to how the alignment process works so if you see good coverage of an amplicon in the amplicon depths plot but mismatching primers in this plot it is more likely to be errors in the consensus sequence alignment.
Sample Reports
Sample reports look like the following screenshot:
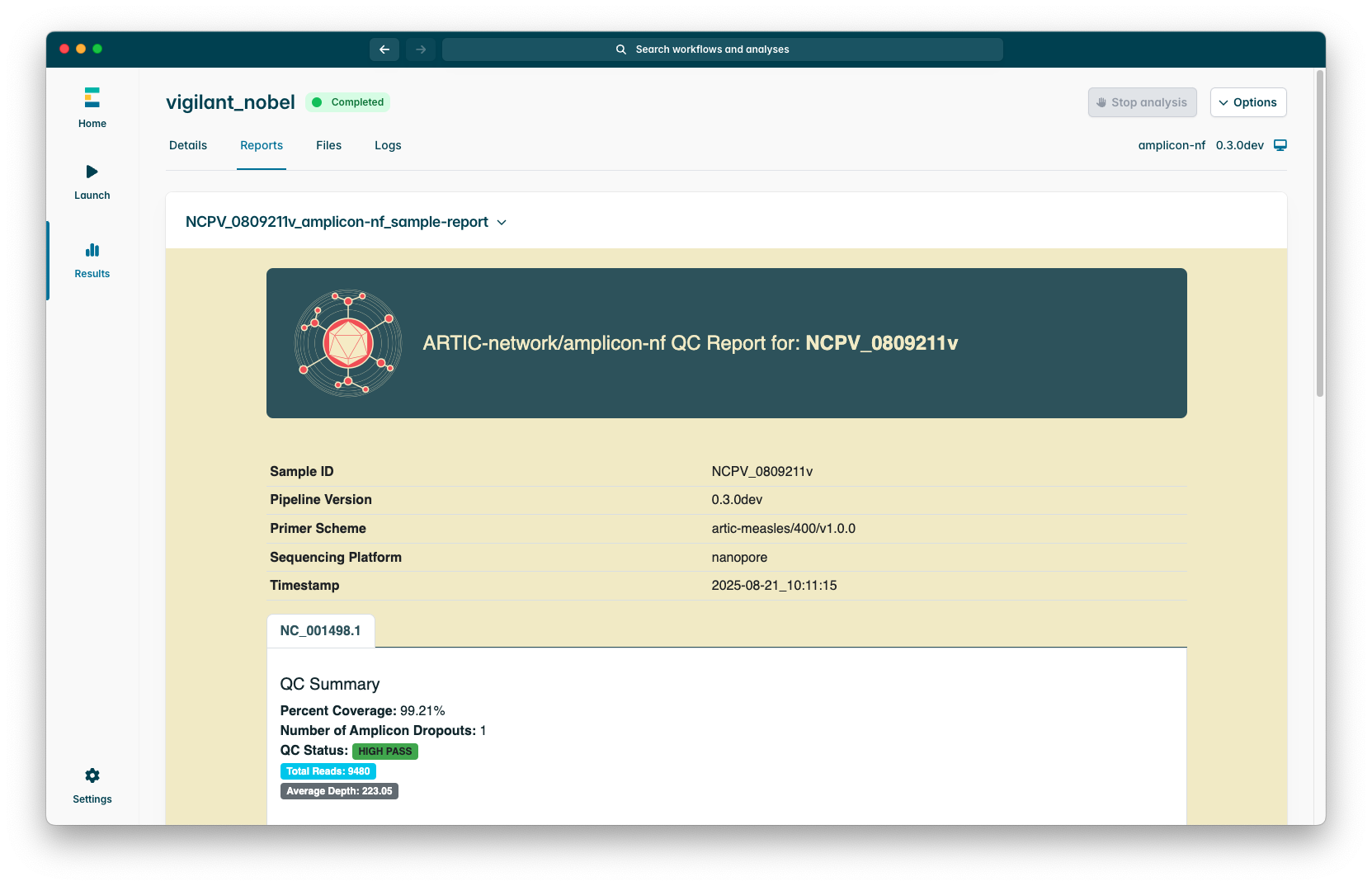
Please note that for segmented viruses (e.g. Influenza) there will be a tab for each segment with separate QC metrics and depth plots.
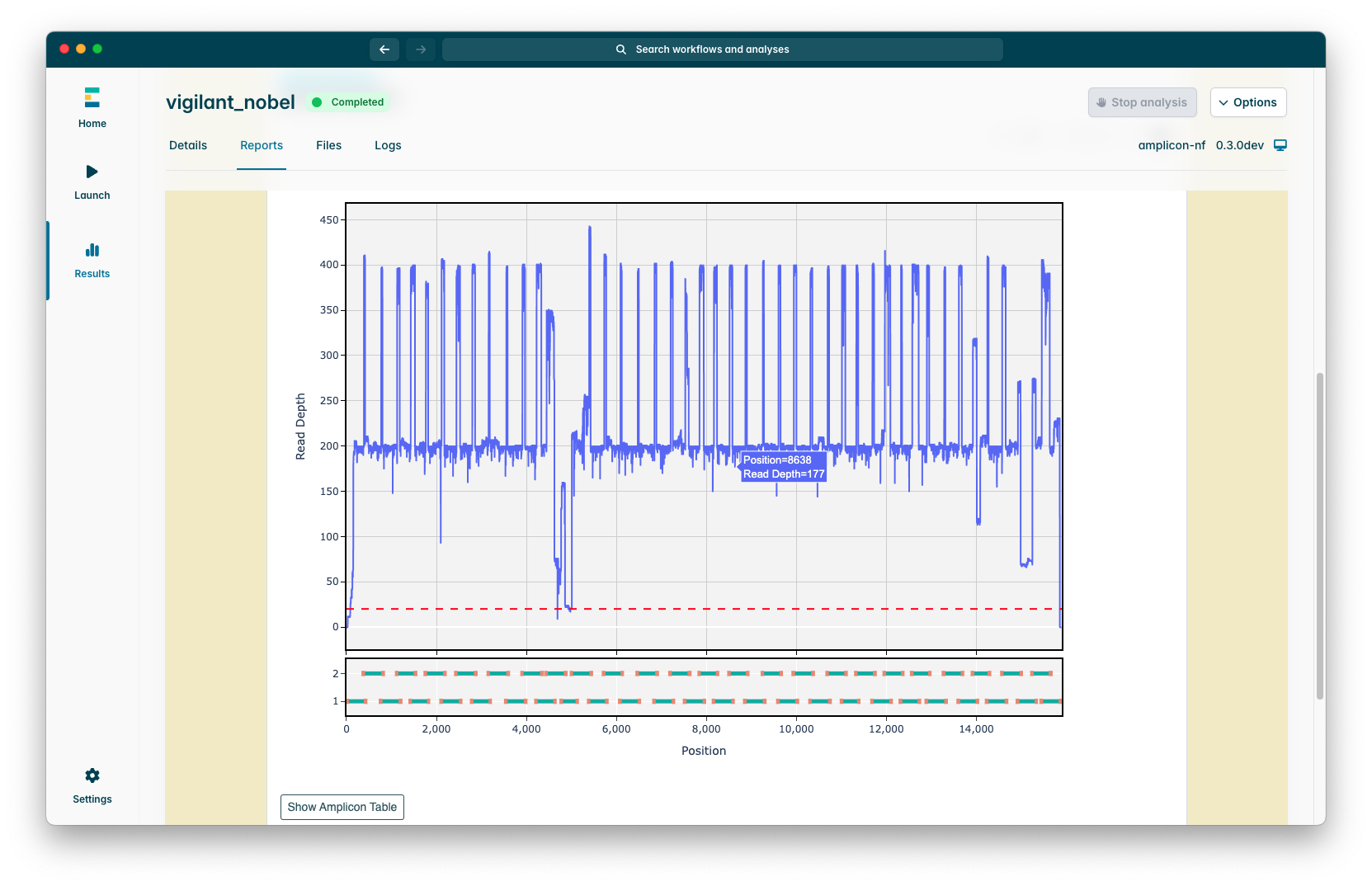
The depth plot is composed of two parts, the upper section indicates read depth across the segment whereas the lower section shows the primer / amplicon positions relative to this, this can be helpful for spotting issues with your data such as amplicon dropouts, samples where a primer pool was not added, etc.
Output Files
The “Files” tab will show you the individual output files within the output directory, as you can see below:
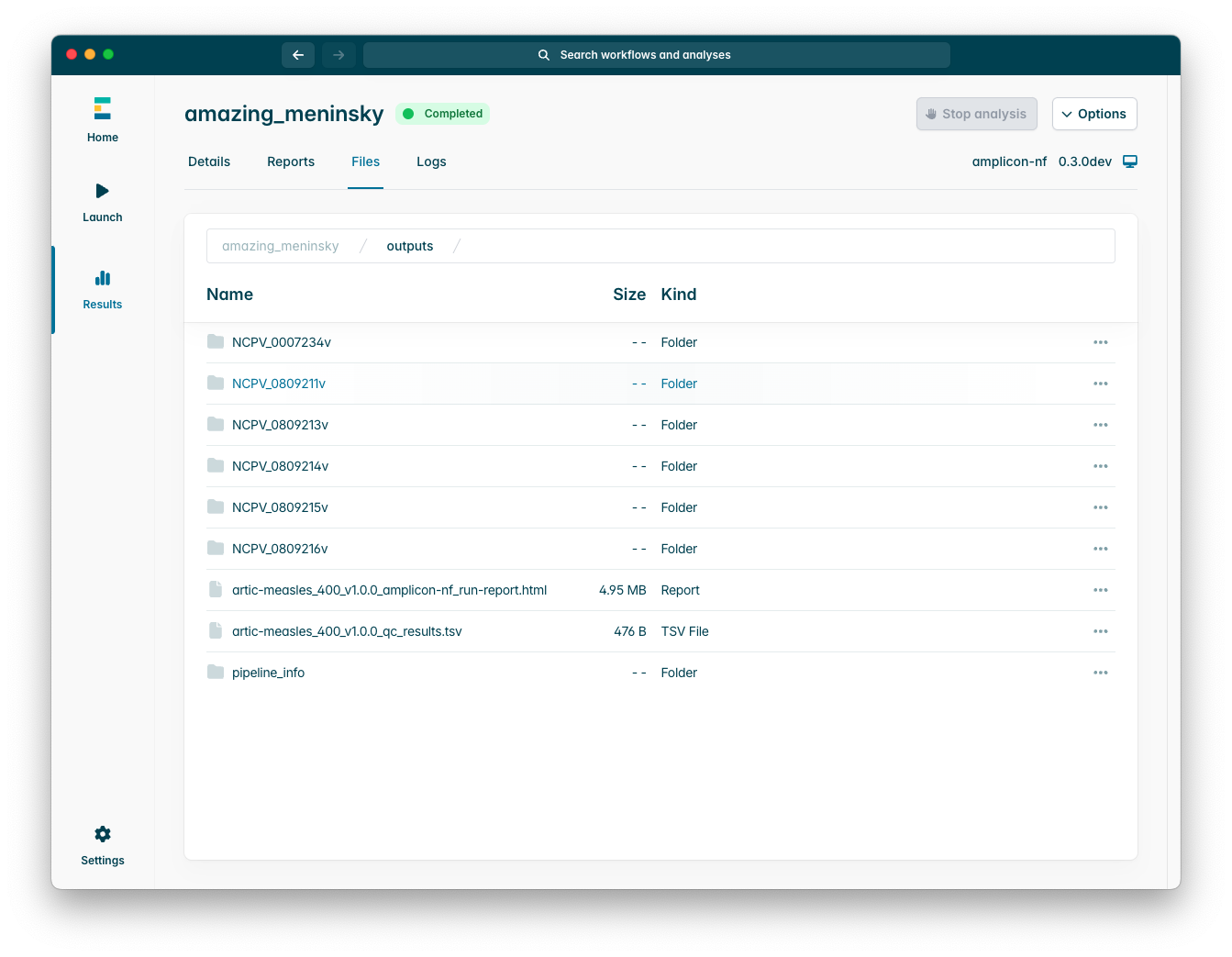
Please note that for each sample (line of your samplesheet) there will be a subfolder containing the specific outputs for that individual sample.
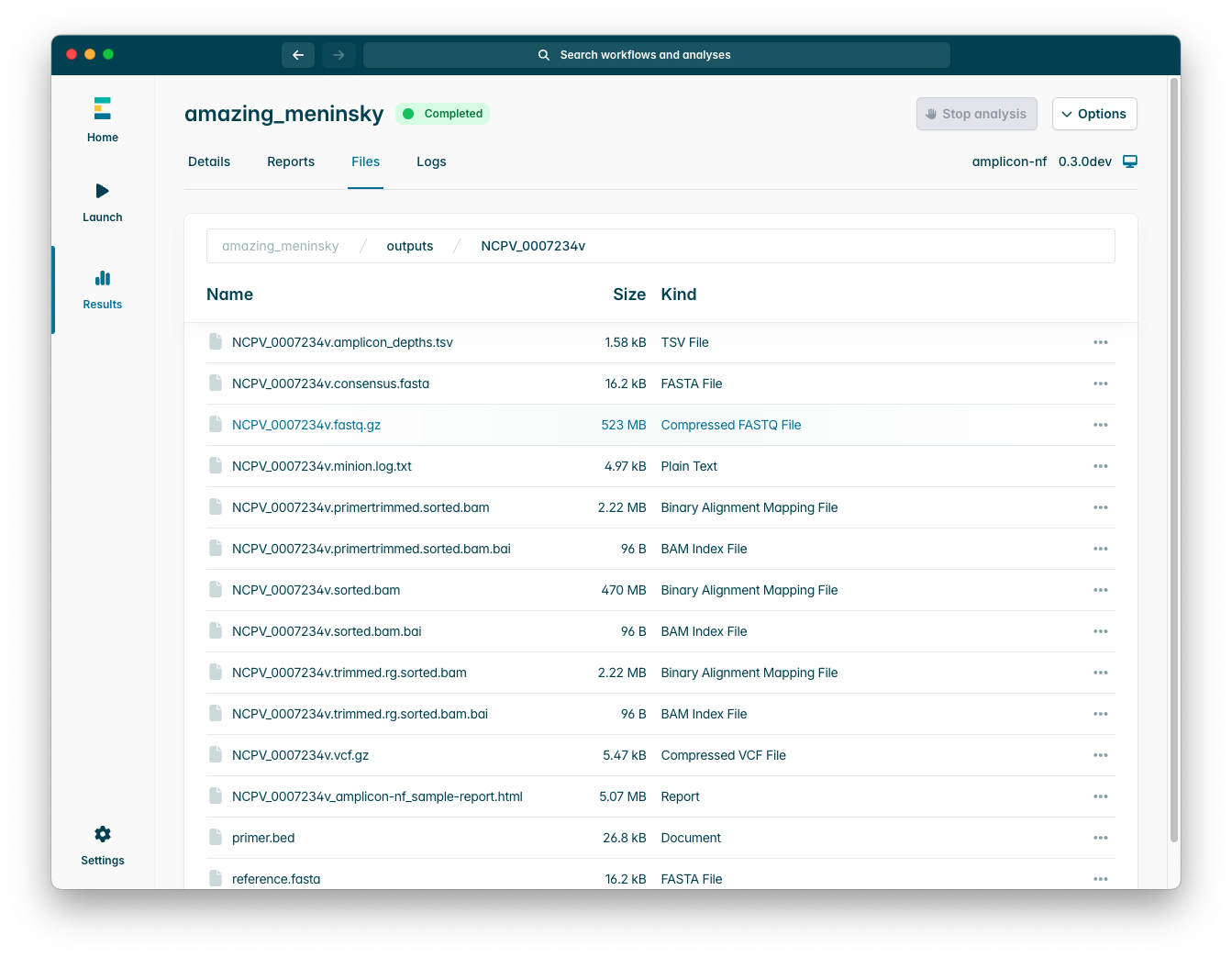
For a more detailed description of the files amplicon-nf produces, please see the amplicon-nf documentation which will always be the definitive source for this.
