Overview: A complete tutorial to combine newly generated consensus genome sequences with background and metadata, running sequence and metadata quality control, multiple sequence alignment, site masking or problematic sequence removal, maximum likelihood phylogenetic inference, and tree quality assessment. The software tools used today include the raccoon toolkit, MAFFT and IQTREE, run through a Nextflow pipeline within the EPI2ME interface. Raccon, MAFFT and IQTREE can be used as command line tools, with full command-line documentation available for raccoon available at artic-network.github.io/raccoon.
Working with multiple FASTA files and creating clear, informative headers for phylogenetic analysis can be much easier with the right tools and guidance. For those who may be less familiar with coding or the command line, tasks such as merging FASTA files or structuring metadata-rich headers can be time-consuming, especially when done manually. This tutorial introduces practical approaches that simplify these steps and help reduce the chance of errors in metadata or sequence entries.
In addition, understanding and critical evaluation of the quality of results is an important part of viral phylogenetics. This tutorial will guide you through sequence quality-control steps, highlight potential issues that can arise during multiple sequence alignment, and help you confidently assess and interpret the resulting phylogenetic trees. An interactive description of the pipeline can be found at artic-network.github.io/raccoon/docs/interactive_pipeline
In this tutorial, the pipeline will be run using the EPI2ME user interface. Users do not need to run every command manually, and do not need knowledge of the command line to run raccoon, however they should understand each stage. Users that are familiar with the command line can run different modules independently in their own custom workflows to aid in post-consensus analysis QC.
Learning outcomes
By the end of the session, participants should be able to:
Explain why metadata harmonisation is essential before phylogenetic analysis.
Identify common metadata and sequence quality problems and describe their downstream impact.
Understand the importance of alignment curation, and the impact of alignment issues on phylogenetic inference.
Appreciate the importance of background data and sampling bias.
Interpret key outputs from raccoon modules seq-qc, aln-qc, and tree-qc.
Critically assess root-to-tip plots, read a phylogenetic tree structure, and understand phylogenetic structure that may indicate upstream analytical issues.
Prerequisites
This tutorial assumes the following software is installed:
Docker
EPI2ME
Setup
1.Today we will run the raccoon-nf pipeline through the EPI2ME user interface. Please first install the EPI2ME desktop application using the provided link. Follow the setup instructions in the package to install and run EPI2ME.
2.Once you have successfully installed, launch EPI2ME.
3.To access EPI2ME without creating an account, click on the three dots at the bottom of the window, and click “Continue as guest”.
4.When you have successfully launched EPI2ME, you should see the above screen. To install the raccoon-nf pipeline, click to open the “Launch” window in the panel on the left hand side.
5.Click on “Import workflow” in the top right of the window, and then “Import from GitHub”.
6.To import the raccoon-nf workflow, paste “https://github.com/Desperate-Dan/raccoon-nf” into the box and click “Download”.
7.You should see the above screen if the workflow installed correctly. Click “Open” to launch the workflow.
1. Understanding and exploring the datafiles
Concepts to cover
FASTA and metadata formats
The content of the provided files
Steps
1.Phylogenetic analysis requires two main types of data: sequence data, in the form of a FASTA file, and metadata, in the form of a TSV (tab separated value) or CSV (comma separated value) file. Often metadata is stored in spreadsheets, such as in Microsoft Excel or Google sheets. If you download a set of sequence data from Pathoplexus, the accompanying metadata is available as a TSV.
2.FASTA files
A FASTA-formatted file contains sequence records, which can be amino acid or nucleotide sequences. A record minimally contains two pieces of information:
The sequence ID (e.g. PP00001)
The sequence itself (e.g. CGATCGAT…ACTGACT)
Format details:
The sequence ID is stored in the header line, denoted by a > symbol
The header line may also contain additional information (sequence description) after the first space
Important: The sequence ID must not contain whitespace (spaces or tabs)
The sequence is stored on the following line(s)
Sequences can be split across multiple lines for readability
The next record does not start until the next line that begins with >
Select which of the following are good/ valid FASTA records:
a)>PP00010 barcode=barcode01 AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACG AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACG AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACG
In order to properly inform our analysis, we need to integrate our sequence data with sequence metadata. Metadata is data that provides additional information about our samples, such as collection date or location. This is usually supplied as an additional file in CSV or TSV format.
Depending on the data collection process, planning, and ethics approvals, metadata may be very detailed or more sparse.
Rank the following types of metadata in order of how useful they may be for genomic epidemiology: Location, immune status,travel history, sample collection date, ct value, symptoms, symptom onset date, gender, age, occupation, patient eye colour, patient height
Which date format should be used as a standard?
Why should we standardise date formats?
4.In this tutorial, you will have two FASTA files: a set of background sequence records, and a set of newly generated consensus sequences that you need to fit into the known diversity and interpret.
We are also providing metadata files to accompany the sequence data. Much of the work in preparing metadata files has already been done, however use these files as a guide for future analyses.
If you have carried out a sequencing run as part of the workshop and have new case data, use that file. Otherwise, a FASTA file of case data can be downloaded from here.
5.Open the input files in a text editor.
What information is provided in 1) the FASTA file and 2) the metadata files?
What column contains the sequence ID?
What column contains the sample date?
What column contains the most detailed location data?
What other information would be useful for our analysis/ interpretation?
2: Understanding raccoon-nf pipeline
Concepts to cover
What steps are run as part of the raccoon-nf pipeline
Pipeline details
Running best-practice phylogenetics can be challenging, however with the raccoon-nf pipeline a simple alignment and phylogenetic workflow can be performed in a single step. The pipeline itself is configurable, however in this tutorial we will be running the steps shown in the figure below.
A) Input files
input sequences (one or more fasta files or directory containing fasta file)
input metadata (one or more metadata files (csv or tsv) or directory containing metadata files)
B) raccoon seq-qc
Outputs:
a combined fasta file with sequence headers harmonised and populated from the metadata fields
seq-qc_report.html (a report describing the dataset, the matching, the output and any issues identified with the data)
seq-qc_filter_failures.csv (sequences that do not pass qc filters, max n and min length)
seq-qc_metadata_issues.csv (flagging missing metadata fields or sequences that failed to match metadata)
C) alignment
Multiple sequence alignment is a key step prior to running phylogenetics. It is the scaffold upon which we can begin to reconstruct the evolutionary relationships between different sequences in the tree. We will run alignment using MAFFT, which is a popular software tool for creating multiple sequence alignments.
Output:
An aligned fasta file
D) raccoon aln-qc
A high-quality alignment is crucial to generating a good phylogenetic tree. Being able to accurately assess whether there are issues with your multiple sequence alignment is a key skill that we will cover today.
The alignment is checked for various issues that may impact the quality of the phylogenetic inference. Different kinds of SNPs (clustered SNPs, N-adjacent SNPs, gap-adjacent SNPs) are flagged that may suggest issues with the alignment or with a given sequence. If a given sequence has many issues flagged (default >20), that sequence is flagged for removal from the analysis. Flagged SNPs do not necessarily mean there is anything wrong with the SNP, it may reflect genuine biological variation. However, these sites may need to be investigated closely.
Output:
aln-qc_report.html (a report describing the input alignment, n content and any SNPs that were flagged as possibly pro)
mask_sites.csv (describes the sites flagged for investigation or masking and the sequences flagged for removal)
E) tree estimation
Tree building is run using IQTREE. The substitution model used is configurable and an outgroup can optionally be included. If an outgroup is included, ancestral state reconstruction will be run during the tree building process to provide additional checks on the tree, and the outgroup sequence will be pruned off from the final tree. In this case, as we are not yet familiar with the data, we will not select an outgroup as it is not clear what an appropriate outgroup would be.
Key output:
*.treefile (a maximum likelihood tree file)
F) raccoon tree-qc
Output:
tree-qc_report.html (report showing the tree, a root to tip and any issues that were flagged during the tree-qc process)
*.phylo_flags.csv
A midpoint rooted tree (if no outgroup provided)
Branch reconstruction file (if outgroup provided)
State difference file (if outgroup provided)
3. Running raccoon-nf in EPI2ME
Concepts to cover
Launching a workflow in EPI2ME
Configuring the raccoon-nf workflow settings to match the input data
Steps
1.To launch an instance of the raccoon-nf workflow, click the “Launch” button.
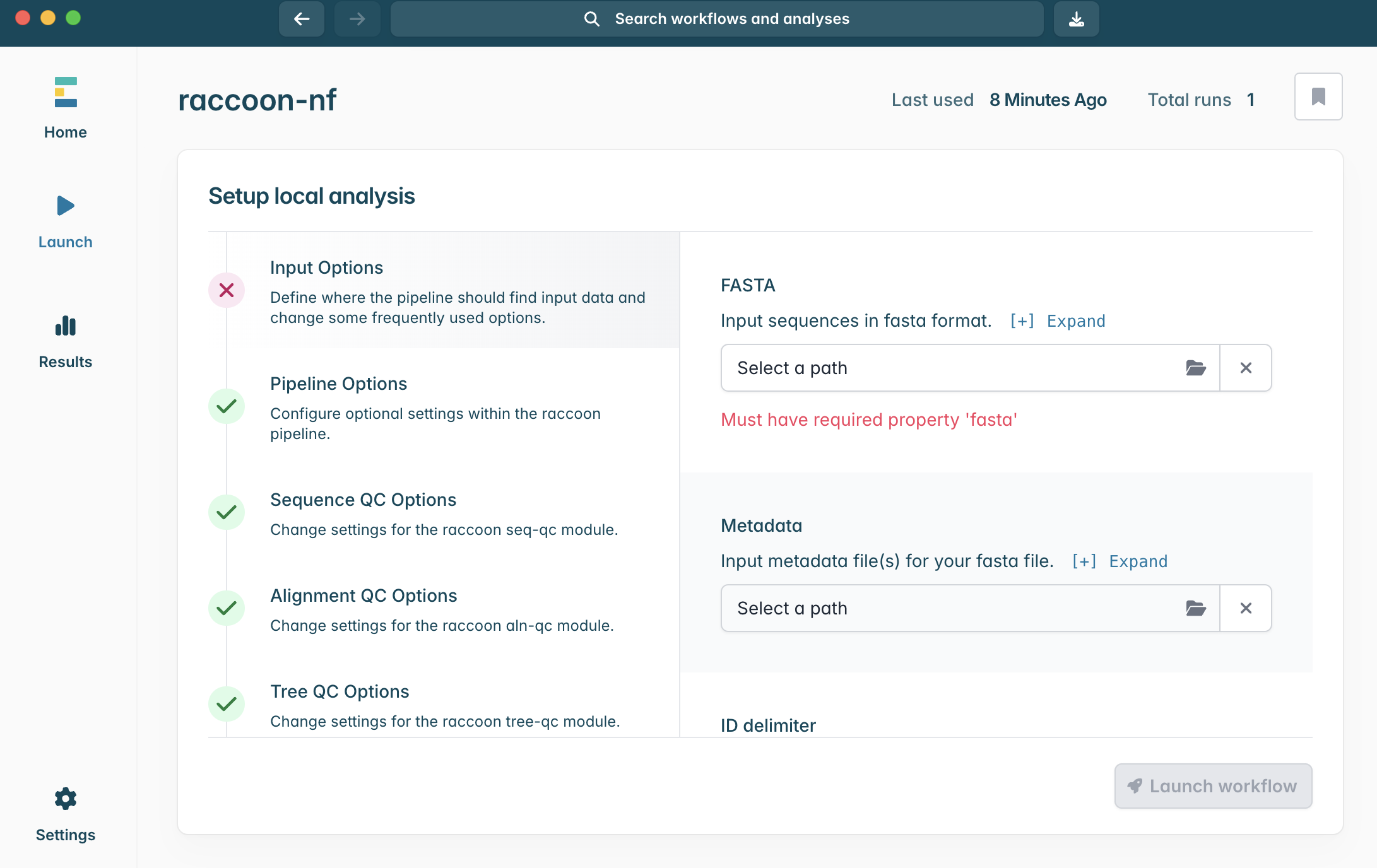
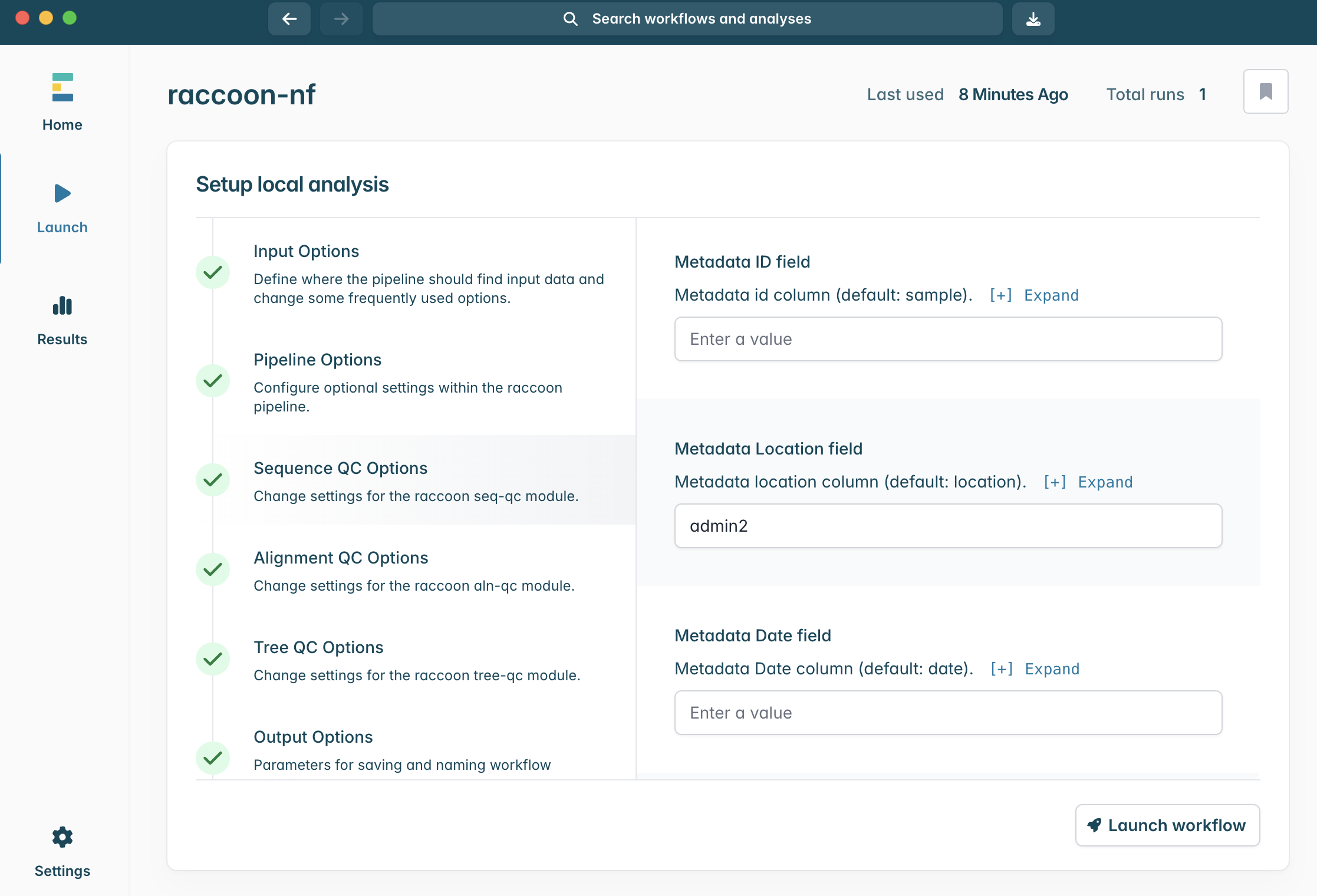
2.This will open the above window, with all the configuration options for the analysis. Minimally, the pipeline requires a FASTA input to run. Today we will run with two FASTA files (background historical data, and the newly sequenced case sequence data) and the two metadata files with a row corresponding to each of the sequence records.
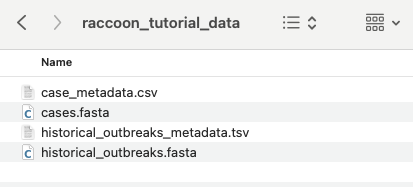
3.If you haven’t already, make sure to download and unzip raccoon_tutorial_data.zip. Drag your newly sequenced case FASTA file to the same directory, as shown above. The directory should now contain four files.
4.In the Input Options panel, select the directory that contains your FASTA files (it should have unzipped into a directory called “raccoon_tutorial_data”), and select the same directory for your metadata files. Raccoon-nf will automatically detect which files present are FASTA files and which ones are metadata files based on the file extensions (i.e. .fasta or .csv/.tsv). We can leave the remaining Input options as default.
5.We will leave pipeline options as default for this tutorial, but feel free to explore the configurable pipeline settings.
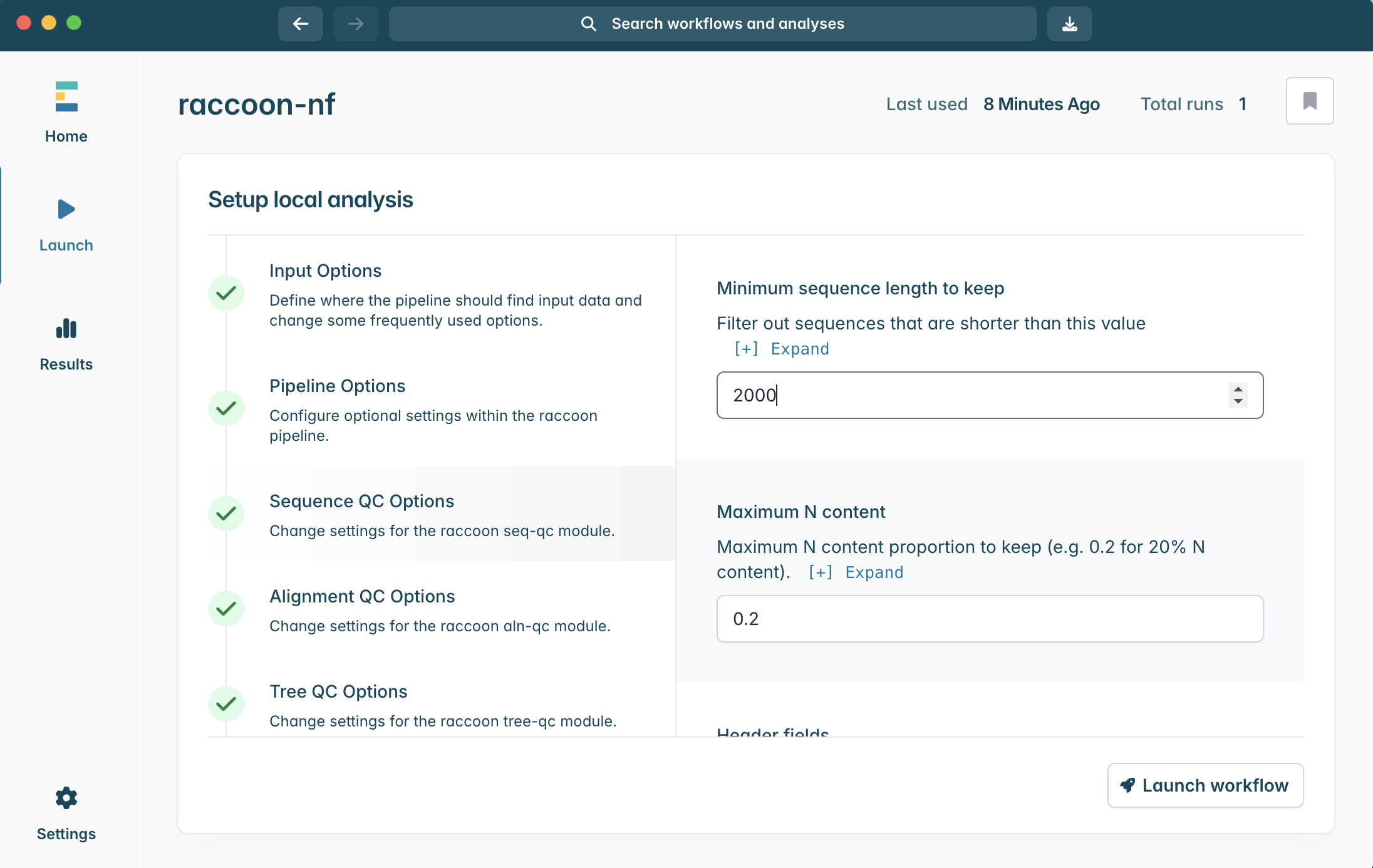
6.Click into Sequence QC options. We can tell you we know the example genome is ~3,200 nucleotide bases in length. When setting up our raccoon-nf run, we want to include genome sequences that are complete, or nearly complete genomes.
What would a sensible minimum sequence length be for this analysis?
What would a good maximum N content be? What are the tradeoffs?
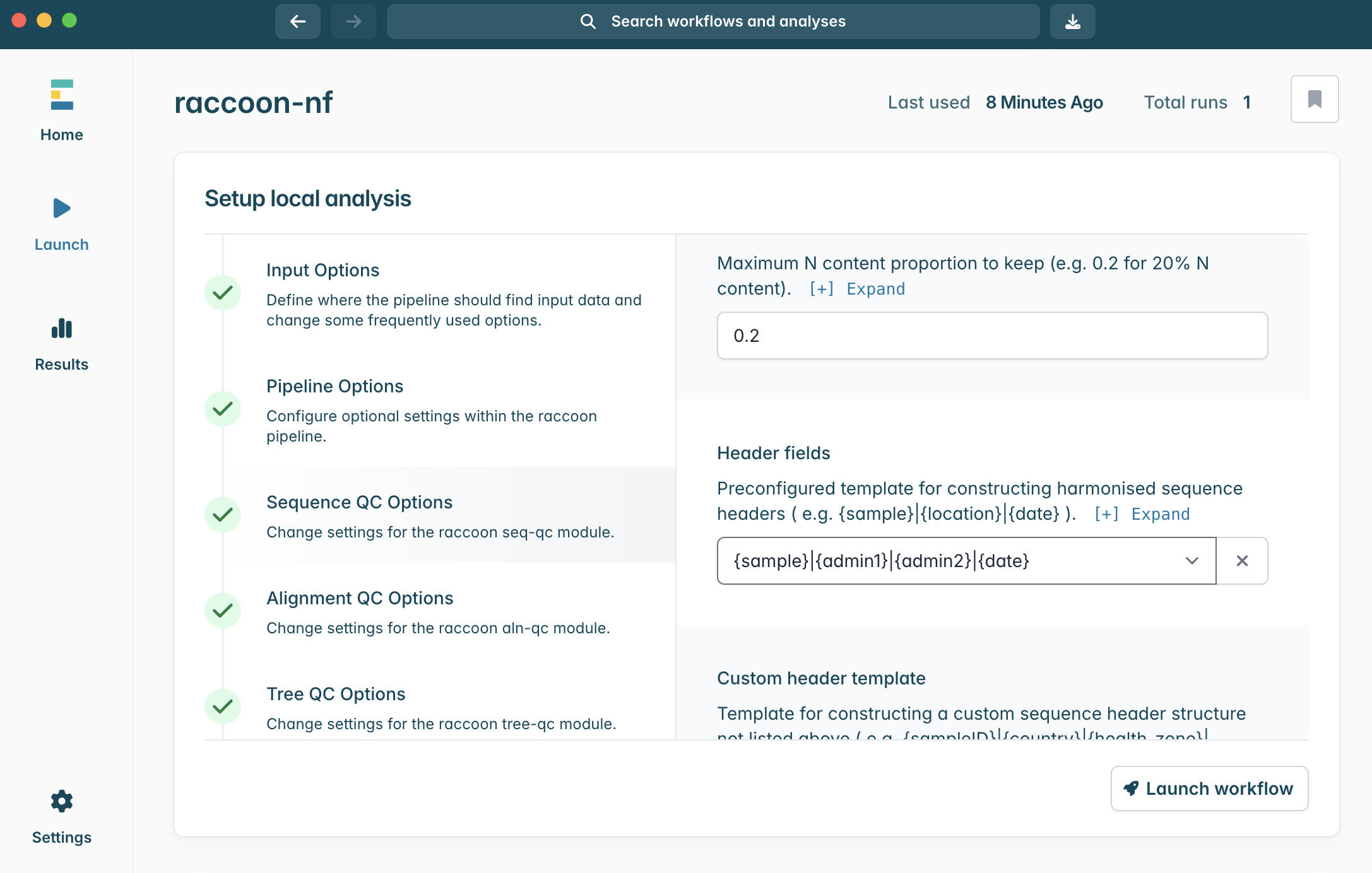
7.Scroll down within Sequence QC options to Header fields. The default header template is {sample}|{location}|{date}.
Think back to our exploration of the provided metadata files. Will this template match up with our metadata?
Tip: look at the column headers
Given three alternative templates below, rank them best → worst and justify:
1. {sample}|{date}
2. {sample}|{admin1}|{admin2}|{date}
3. {admin2}|{date}
4. {sample}|{location}|{date}
8.Scroll down in Sequence QC options. The default Metadata ID field is sample. Write in the location column we would like to have raccoon use.
Between admin1 and admin2, which is the better choice?
What would impact this choice?
Tip: think of how complete metadata may be
9.We will leave Alignment QC Options, Tree QC Options, Output Options and Nextflow configuration as default. Feel free to explore configuration options.
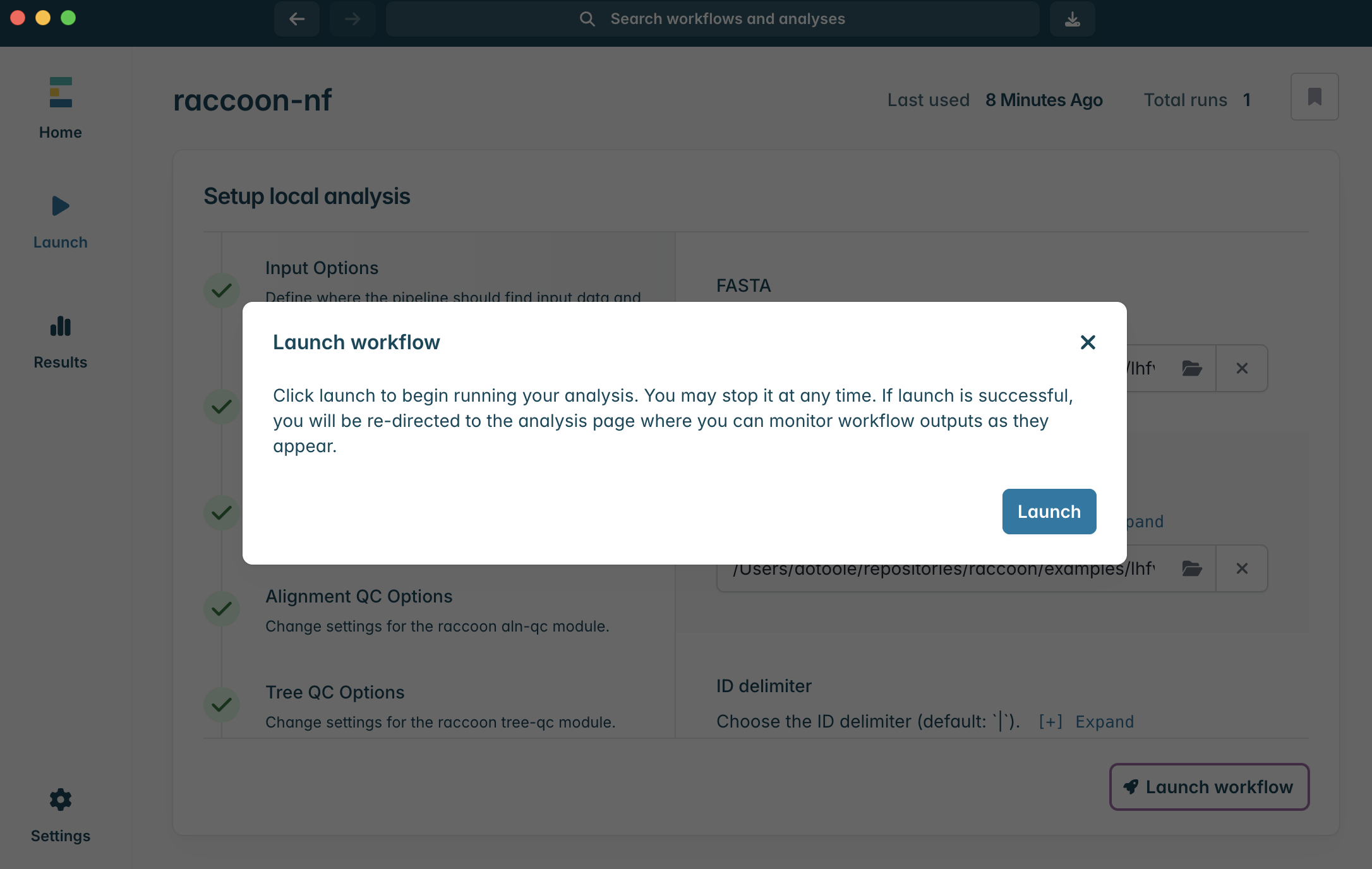
10.To Launch the workflow, click Launch workflow in the bottom right corner. A window should pop up. Click Launch to start the pipeline.
11.The pipeline will begin running and you can monitor the progress of each step.
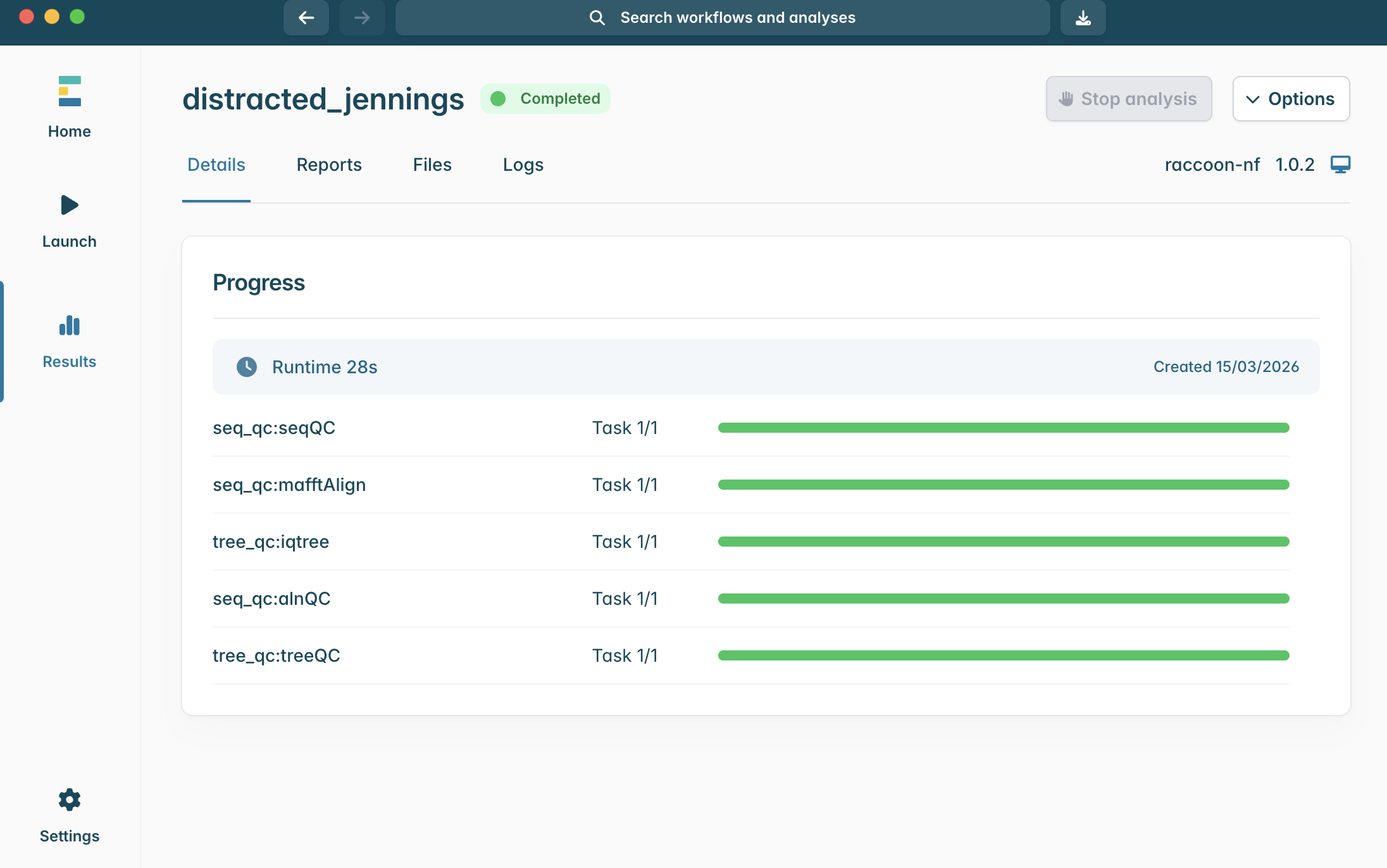
12.When the pipeline successfully runs all steps, you will see the progress status change from Running to Completed.
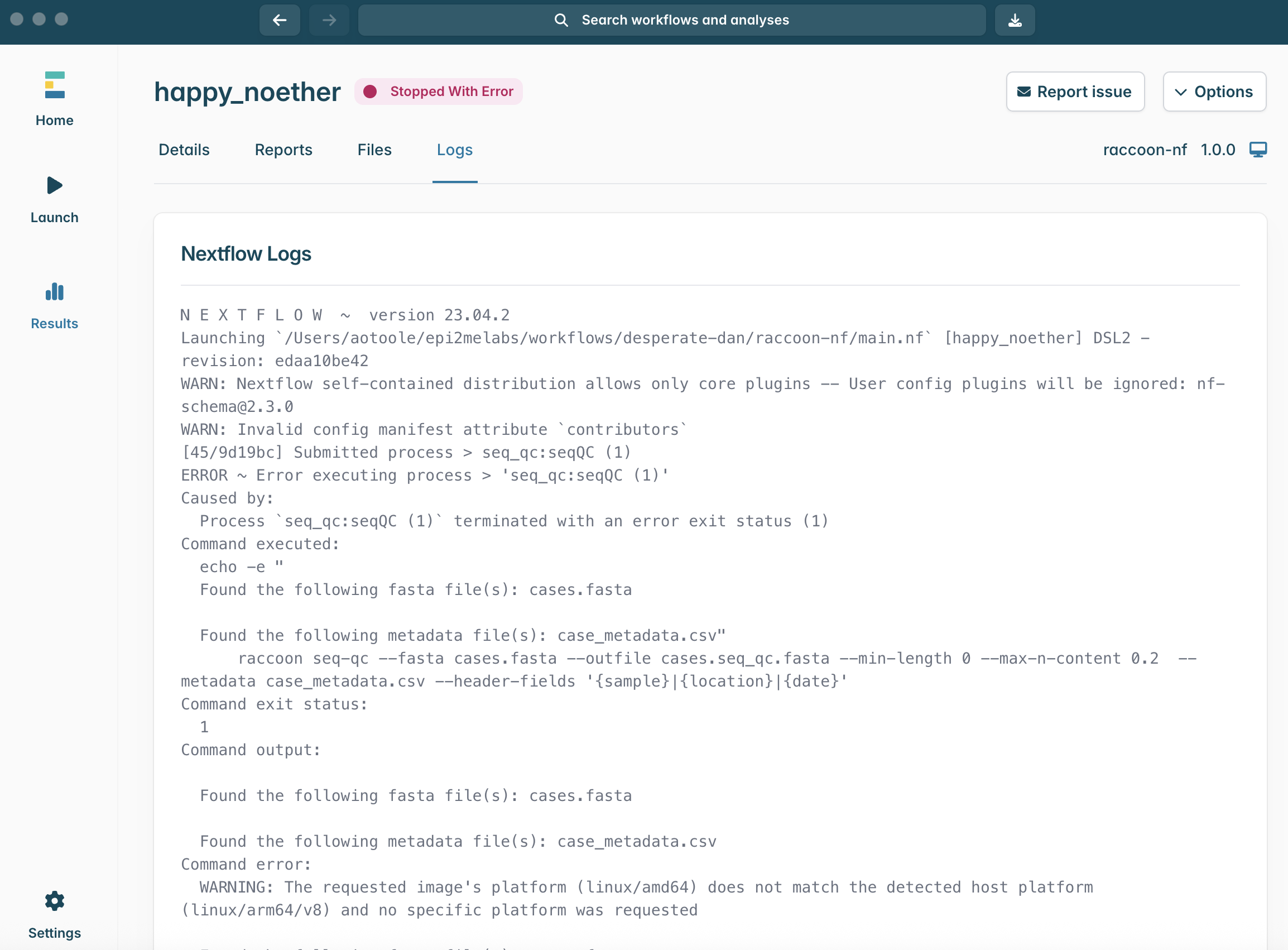
13.If the Running status does not change to Completed and instead turns red and changes to Stopped With Error, something has gone wrong. The ability to read and interpret error messages is the most useful skill for any bioinformatician. If you see this message, navigate to the Logs menu.
14.The Logs window shows the output from Nextflow as it runs and any error messages will show at the bottom. This log can look intimidating, but it contains useful information! If you have an error, scroll down to the bottom. Can you identify the error message?
Tip: it often is printed following the word ERROR
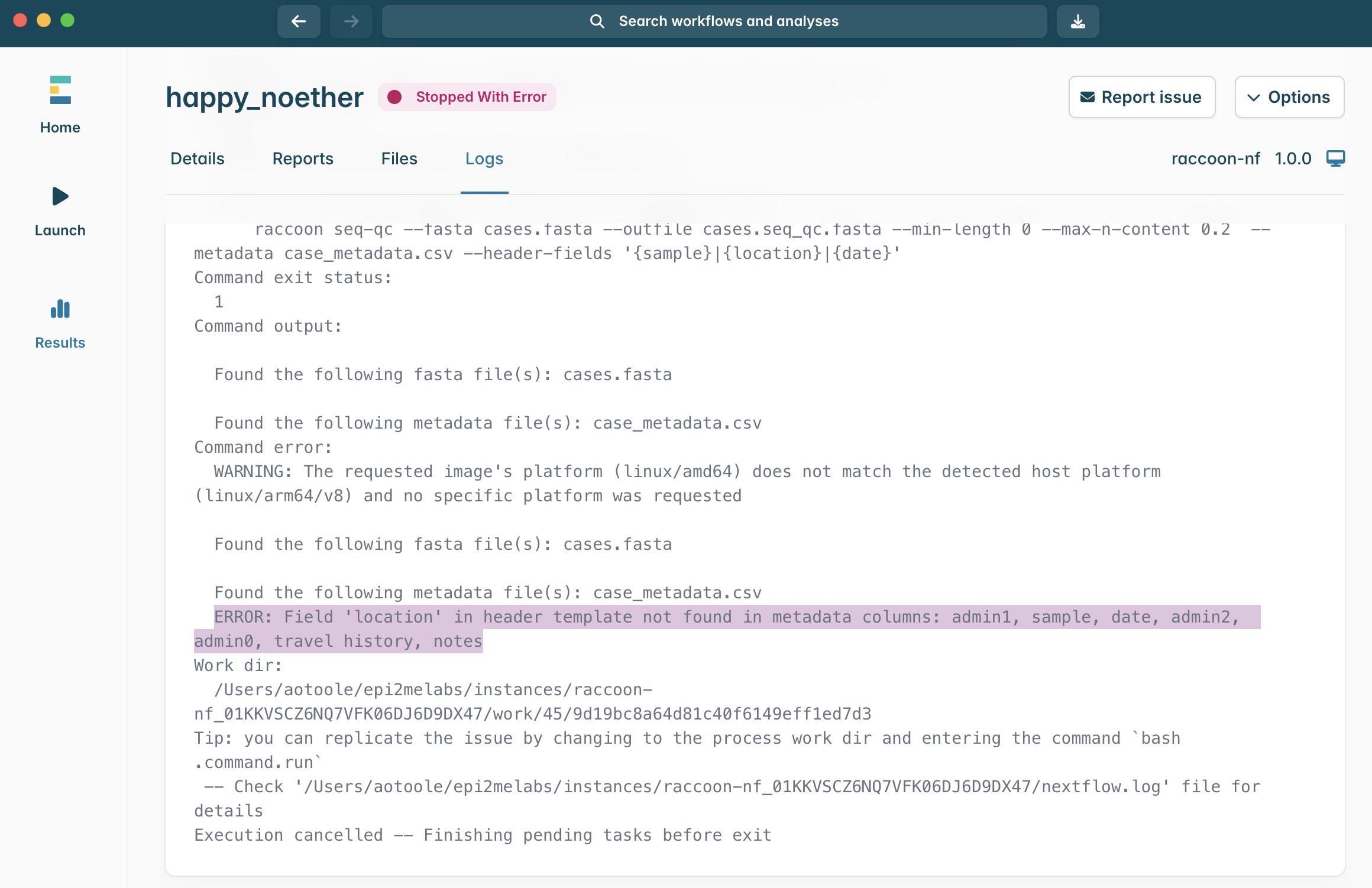
15.In this example, the error reads: ERROR: Field location in header template not found in metadata columns: admin1, sample, date, admin2, admin0, travel history, notes
What does that suggest went wrong during this pipeline run?
How would you solve this error when you run the pipeline next time?
Checkpoint questions:
What criteria are used to filter sequences?
If our metadata file contained the following columns – ID, country, health_zone, sample_date – what would an appropriate header fields template be?
Where can you find information that can help explain an error?
4. Interpreting the output of raccoon-nf (seq-qc)
Concepts to cover
ID matching between sequence headers and metadata table rows
Harmonising metadata from multiple files
Preserving epidemiologically useful fields in headers
Steps
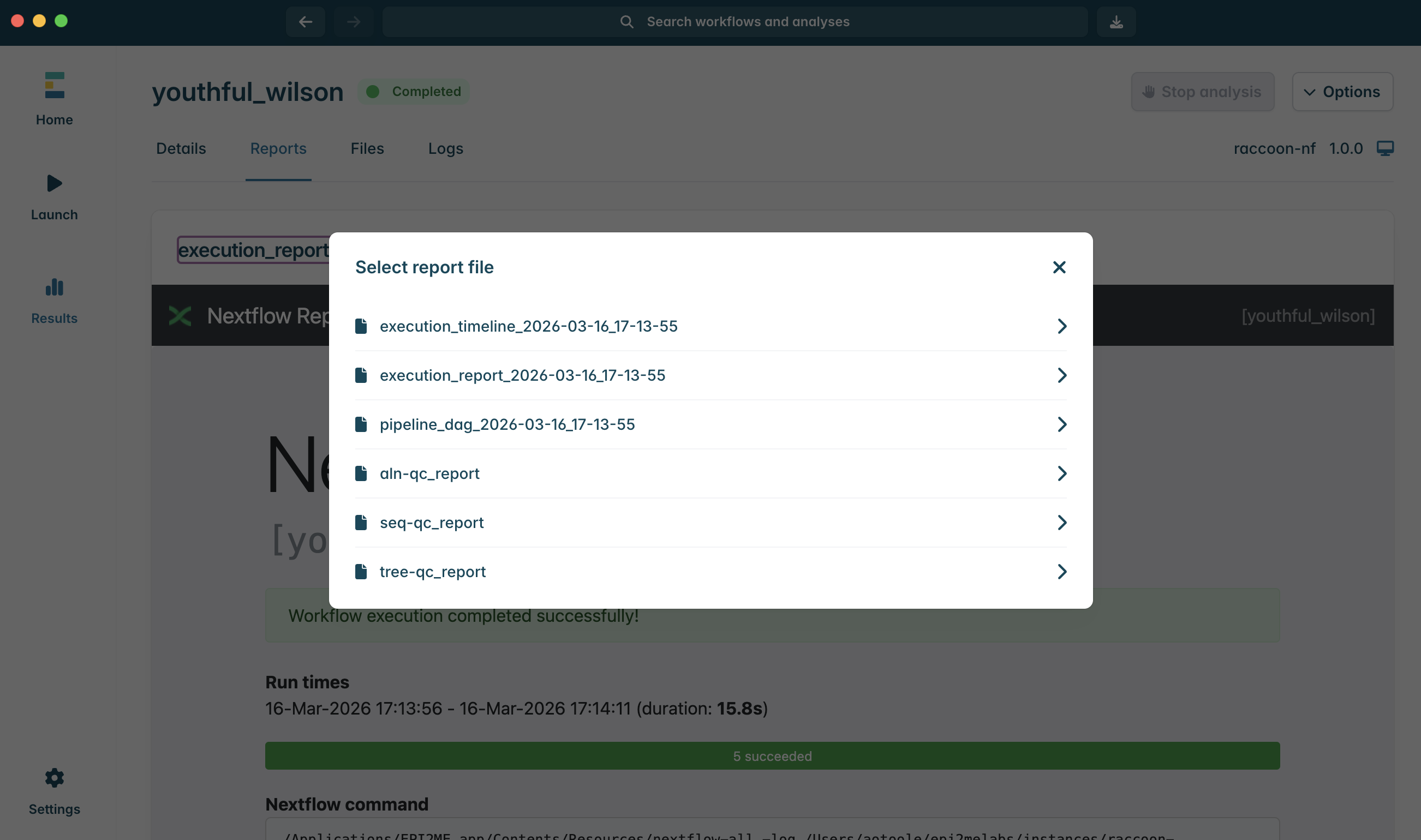
1.Navigate to the Reports tab.
2.Click on execution_report…. dropdown to view the available reports. Select seq-qc_report.html to open the seq-qc report.
3.Explore the seq-qc sections in turn.
The report summary section provides an overview of the data including total sequences, the date range associated with the sequence metadata and any filters applied.
How many sequences were detected in the two files?
The inputs files section describes the FASTA and metadata files loaded into raccoon, with an overview of the number, length and ambiguous base content of the sequences in the input FASTA files. Click to expand each file to view QC information for each sequence.
The sequence length distribution plot shows lengths of the sequences in the input FASTA files.
Are there any outliers in terms of sequence length?
The metadata description plot summarises the geographical and temporal distribution of the dataset provided.
When was the earliest sequence sampled?
How many unique admin2 locations are included in the dataset?
The filtered sequences table highlights sequences flagged for filtering during the QC check.
Were there any sequences filtered out? Why were they filtered?
What is the shortest sequence? Why do we not want to include a short sequence?
What sequence has the highest N content?
The final dataset table describes the final, combined dataset, with harmonised headers.
How many sequences are included in the final dataset?
Finally, any metadata issues such as missing fields or inability to match sequences are flagged in the metadata issues table.
Are there any sequences with missing metadata fields? How has raccoon handled these?
Discussion
Is it better to relax --max-n-content or drop marginal sequences? Why?
What are the risks of including poor-quality but epidemiologically important samples?
4. Interpreting the output of raccoon-nf (aln-qc)
Concepts to cover
The impact of high N content on downstream phylogenetic inference
The importance of visually inspecting your alignment
Steps
1.Next, open the aln-qc report by clicking on the html dropdown. Explore the different sections. Prior to any phylogenetic analysis, it is necessary to inspect the input alignment to assess its quality. Raccoon can help highlight parts of the alignment to investigate further. This is an important step before we start to build our tree, since alignment issues can significantly change the tree structure and our confidence in our conclusions.
2.The summary provides an overview of the multiple sequence alignment, including the number of sequences, alignment length and N content.
3.The alignment N-content section provides a more detailed picture of what parts of the alignment contain ambiguous bases.
Are there long stretches of Ns in our sequence?
What effect might this have on our downstream analysis?
4.If specific sites have been identified by raccoon as possibly problematic, they will be reported in the flagged sites section. The table shows sites identified, sequences they were found in, and their location. The note column explains why each site was flagged.
How many sites have been flagged?
Why were they identified as problematic? What biological vs technical explanation is plausible?
If more than one sequence contains the flagged SNP, is this SNP more or less likely to be biological?
5.These sites are not necessarily problematic, but investigate the alignment quality. These are good places to visually inspect. If a sequence has more than 20 sites flagged, that sequence will be flagged for removal.
Have any sequences been flagged for removal?
6.The diversity plot allows us to see how conserved or variable sites are across the alignment, possibly flagging alignment issues.
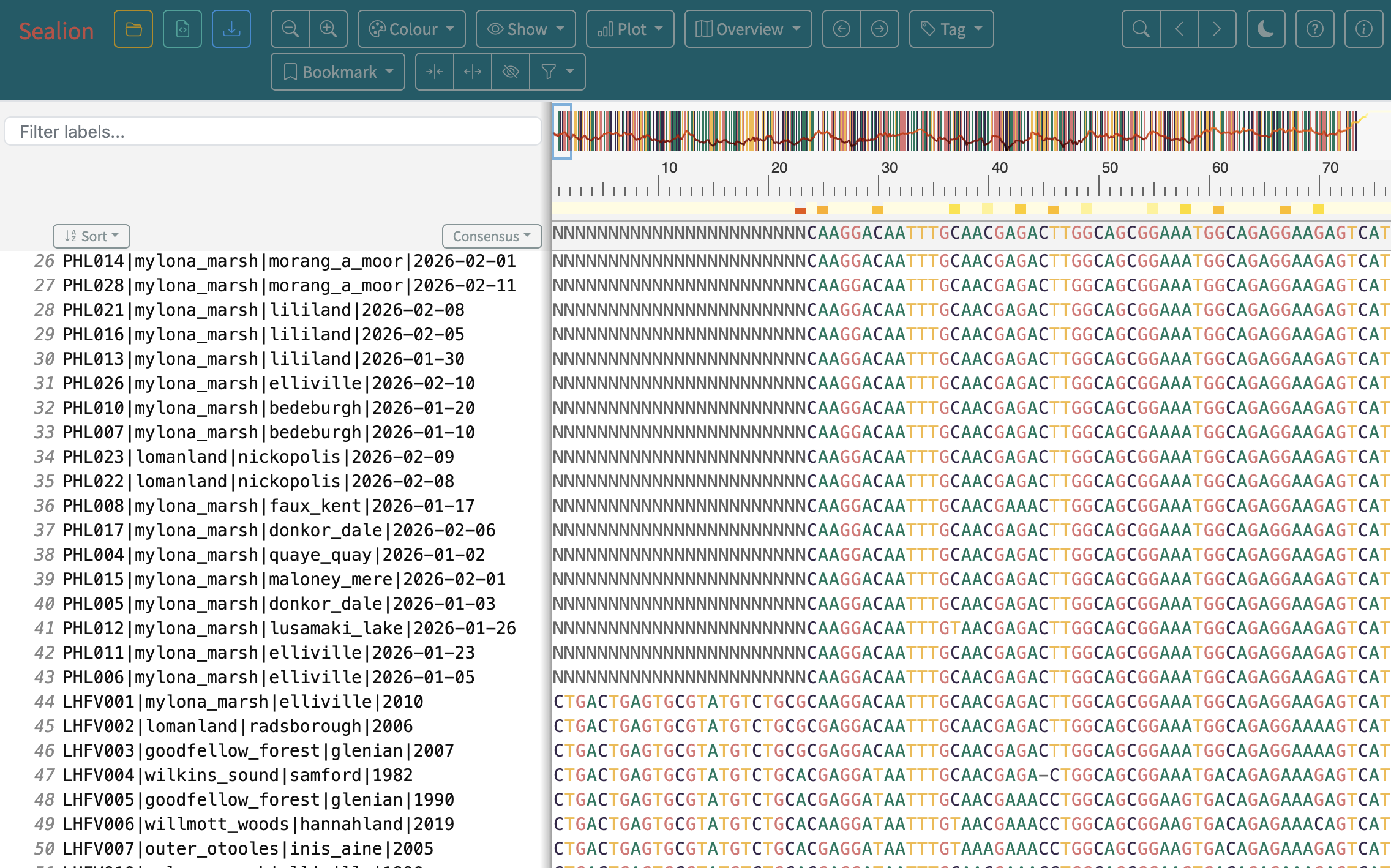
7.Next we will open the alignment file in Sealion to investigate the flagged sites. In EPI2ME, navigate to the Files tab. Then click into raccoon_tutorial_data > then mafft to see the aligned FASTA file.
8.To open the directory to access the alignment file, click the three dots and select Open in Finder or Open folder, depending on your platform. A file browser window will show where the alignment file is located.
9.In a web browser window, navigate to artic-network.github.io/sealion/. Drag and drop your alignment file in to explore your alignment.
10.Use the flagged sites as a guide to assess the quality of your alignment.
Notice the Ns present at the start and end of the alignment in the latest sequence data. What might give rise to this?
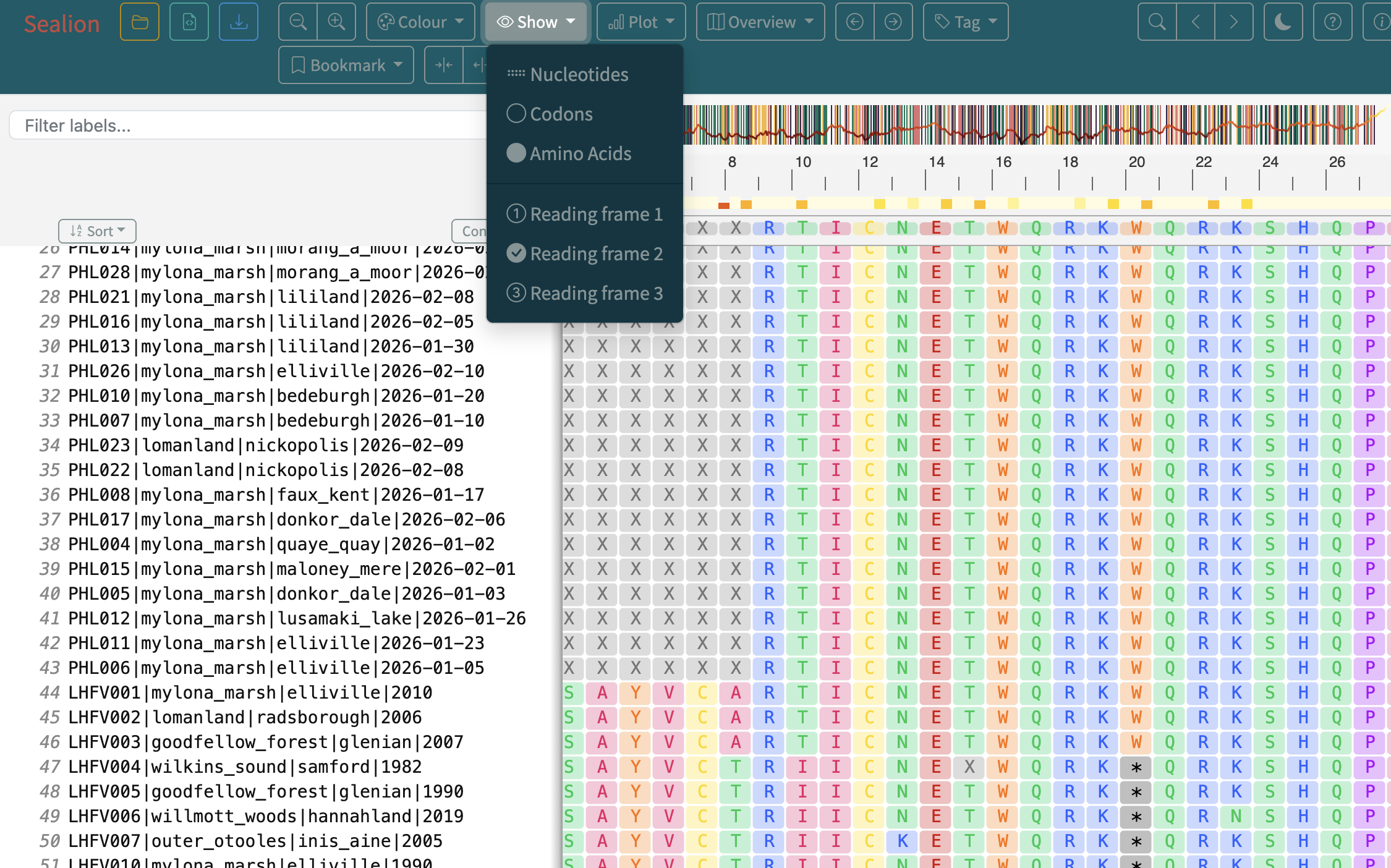
11.Sealion can be used to view amino acid translation of coding sequences in different reading frames (Click Show > Amino Acids). Explore the different reading frames to investigate what the genome codes for.
Can you decipher the code?
4. Interpreting the output of raccoon-nf (tree-qc)
Concepts to cover
How to read a phylogenetic tree
Interpreting a root-to-tip plot
Steps
1.Once the tree has been built, raccoon-nf conducts further QC checks and produces a report. Open the tree-qc_report.html in the EPI2ME browser. The summary provides overview information about the tree. Navigate to the Interactive tree section. Tips can be colored according to various traits.
2.Colour the tips by year, then colour the tips by location.
Do the samples cluster by location and time?
What is the closest historical sequence to our newly generated sequences?
What does the historical context add to the interpretation, and what conclusions would be different if only the case sequences were analysed?
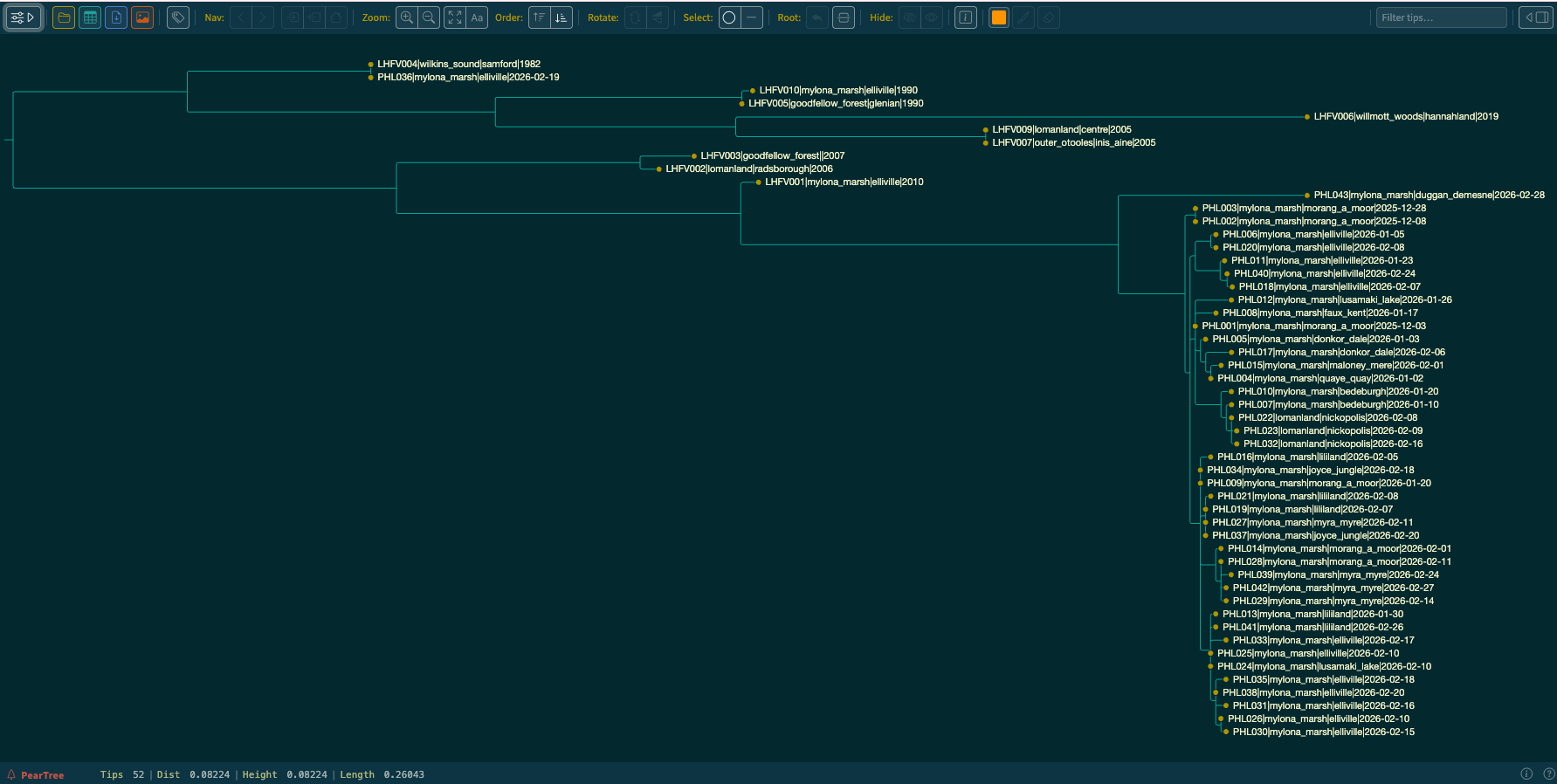
3.Open and view this phylogeny in PearTree.
Can you locate the treefile in the output Files? Refer to the instructions for finding the alignment file.
Tip: look under tree and identify the .treefile file.
4.Inspect the phylogeny, click the visual settings button in the top right corner to open the panel, and turn on tip labels to show names and explore where the newly generated sequences lie within the known diversity.
Do the samples cluster by location and time?
Are all newly generated sequences clustering together?
Describe what the phylogeny tells us about sample PHL036?
Tip: (In the top right text box, type in PHL036 to locate the tip)
What does the phylogeny tell us about sample PHL043?
5.Compare the epidemiological information provided in the metadata with the structure of the phylogeny.
Are the clusters consistent with the epi-links?
6.The Root-to-tip regression plots each tip according to its sampling date and its distance from the root of the tree. The regression line is also plotted, estimating the overall molecular clock of the dataset.
Do most of the recent samples fall along the regression line?
Are there any outliers? What does the root-to-tip plot say about sample PHL036?
What possible causes could give rise to this?
7.If an outgroup had been selected in our raccoon-nf run, ancestral state reconstruction would have been performed and additional QC checks. We have not performed this analysis in this tutorial.
8.Now that we have explored the output phylogeny, there are next steps we could take. We have identified outliers and unrelated cases which we would want to remove prior to running further temporal analysis using Bayesian methods in software such as BEAST. We will not be running temporal analysis today, but you are now in a good position to do additional downstream analysis.
Discussion
What challenges did you encounter during the workflow? Did you encounter any errors during execution?
How would you adapt this workflow for a different virus or dataset?
Tip: Think of read length, header fields and sourcing background data.