Background
The ongoing outbreak of Ebola virus in the Democratic Republic of Congo has highlighted the need for rapid sequencing ability to help with source attribution and aid epidemiological investigations (including environmental reservoirs).
During real-time sequence surveillance of Ebola in Guinea and Sierra Leone in 2015-2016 we employed a PCR amplicon schemes specifically targeting the Makona strain of Ebola. Because this scheme was designed against Makona, we expect that this scheme may not be optimal due to sequence diversity.
Therefore we devised a new scheme using all available Ebola virus sequences and then sequenced Zaire Ebolavirus RNAs at Public Health England, Porton Down. We selected viruses from three separate outbreaks in order to gain confidence in the scheme’s ability to sequence divergent lineages.
This validation acted as a test of our recently published standard operating procedure.
Primer pairs
Sequence coverage (log scale) at 30 minute and 2 hour time points for three different trains:
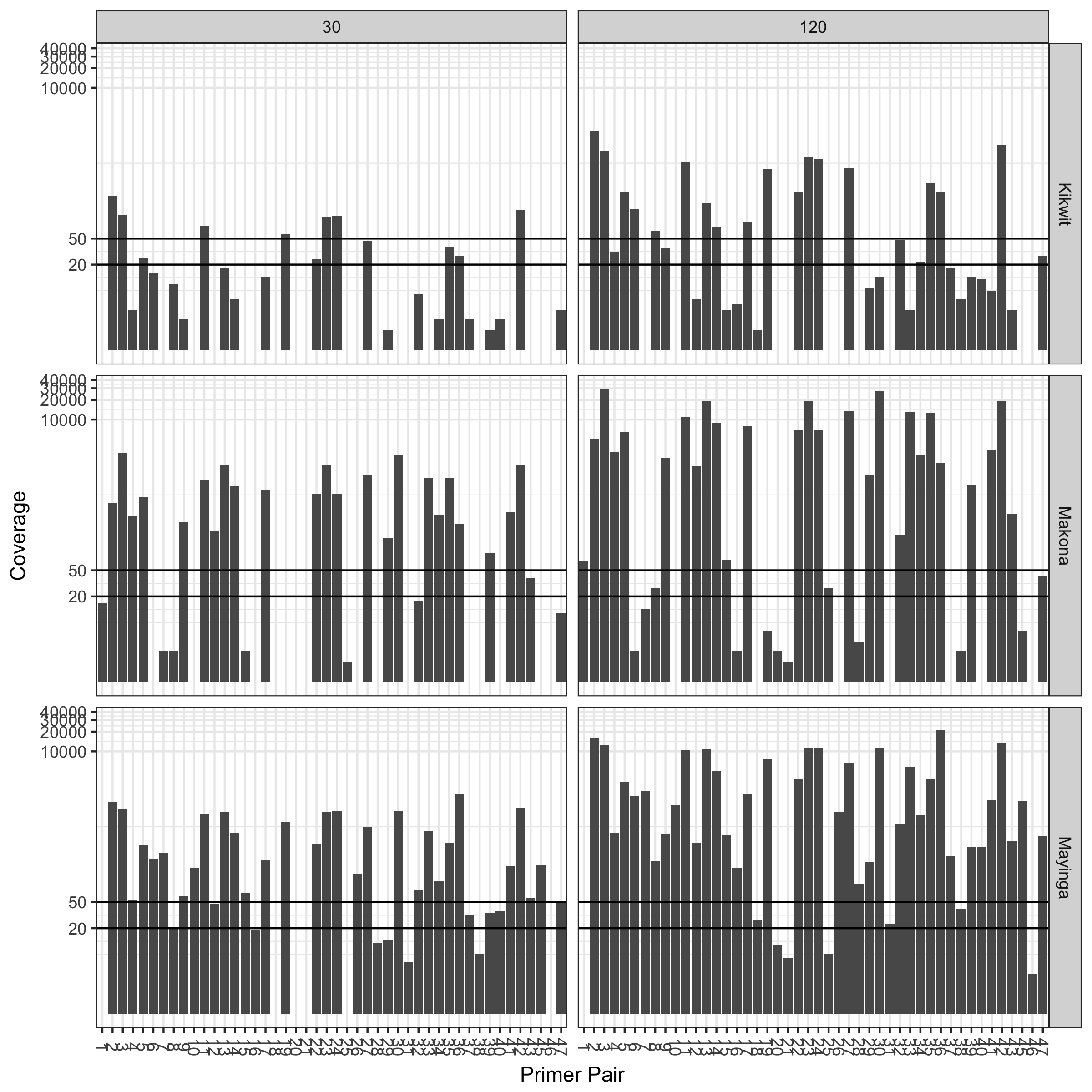
Results
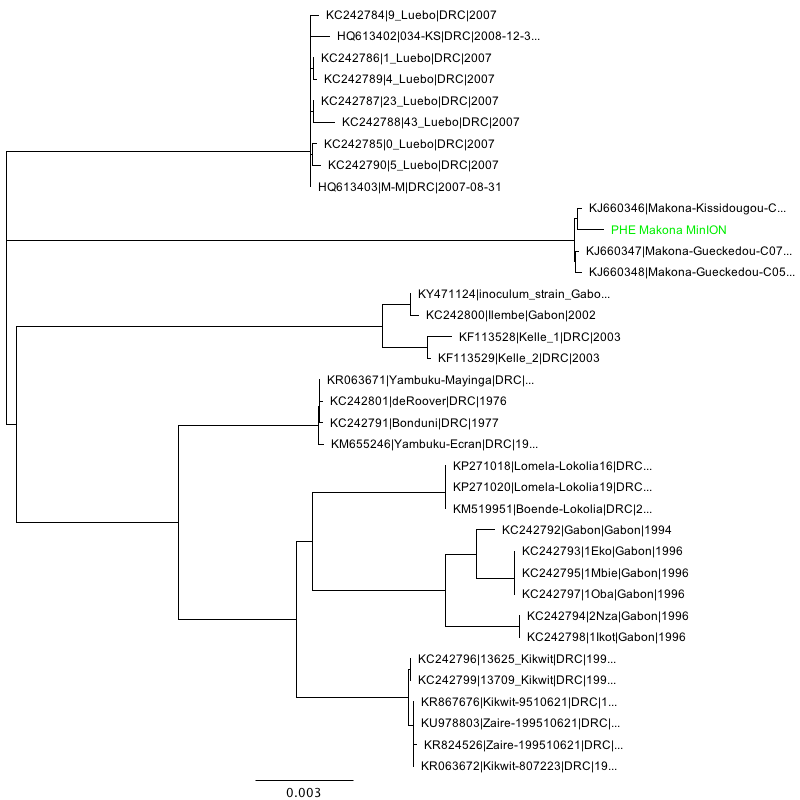
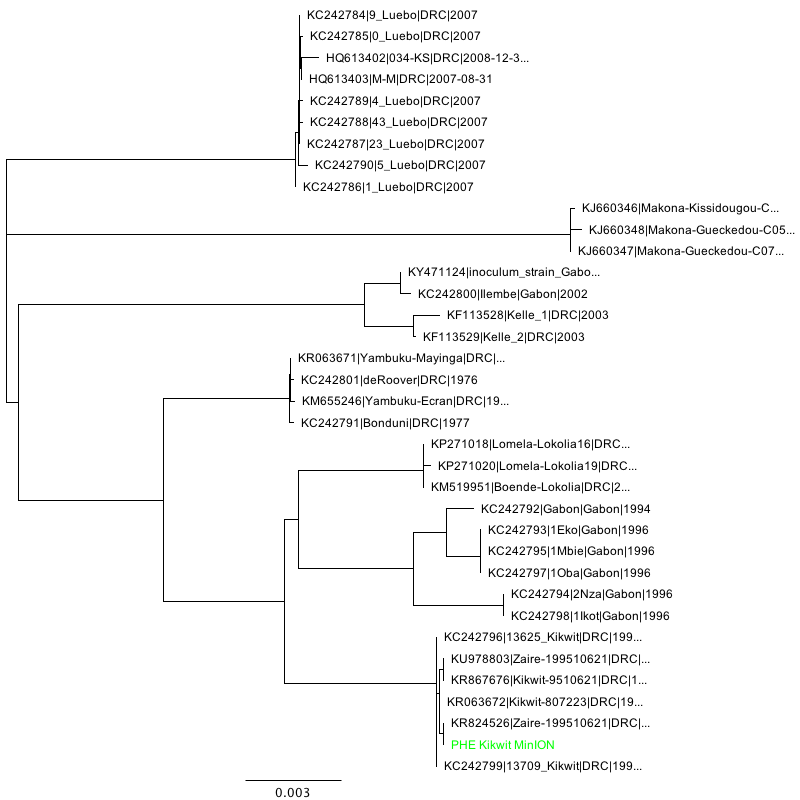
Phylogenetic tree inference (NJ, HKY performed using Geneious 11) of the three consensus sequences in context:
Mayinga strain
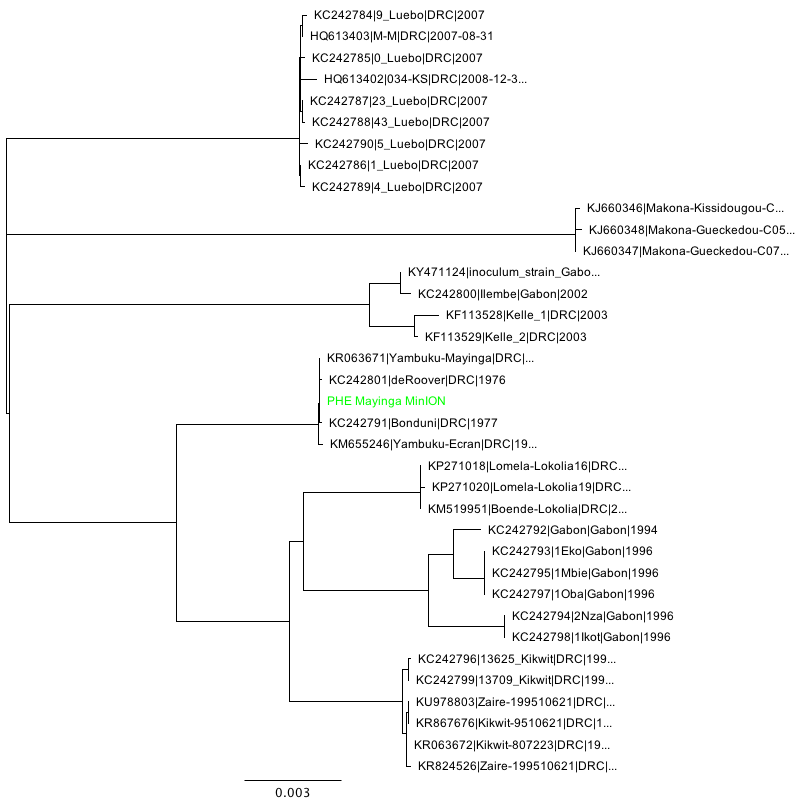
Makona strain
Kikwit strain
Consensus sequences for 2 hour run:
/artic/ebov-consensus-2h.fasta
Data availability
Data hosting provided by MRC CLIMB.
30 minute bulk file:
2 hour bulk file:
FAST5 reads from entire run (~6.4M reads):
artic/fast5/ZEBOV_3Samples_NB.tar [153Gb]
Guppy basecalled (on MinIT) from entire run:
artic/ZEBOV_3Samples_NB_MinIT_guppy.tgz
Credits
- Babak Afrough (Public Health England) for sample preparation, and laboratory and bioinformatics assistance
- Luke Meredith (University of Cambridge) for sequencing library preparation
Acknowledgements
We are grateful to Miles Carroll, Steve Pullan, Richard Vipond and Roger Hewson (Public Health England) for assistance with this validation project and Ben Gannon (RST/Public Health England) for supplying the flowcell.