amplicon-nf: Running the pipeline in EPI2ME
Using EPI2ME for running the ARTIC amplicon-nf pipeline without the commandline
| Document: | ARTIC-amplicon_nf-epi2me-SOP-v1.0.0 |
| Creation Date: | 2025-08-21 |
| Author: | Sam Wilkinson |
| Licence: | Creative Commons Attribution 4.0 International License |
This SOP assumes that you have installed and configured the EPI2ME client, if you have not please see the epi2me desktop client setup guide for instructions to do so!
Generating your amplicon-nf input samplesheet
Before you we begin you will need to create a samplesheet in CSV (comma separated values) format that describes your sequencing run. The contents of this file will be different depending on 1) which sequencing platform you are using and 2) whether you are using a custom scheme.
If you already have a valid samplesheet prepared, you may skip to the next section!
1) Primer Scheme
Whichever platform was used to generate the data, you will need to tell the pipeline which primer scheme you used to generate your data, there are two ways to do this depending on whether you used an official primer scheme (it is stored in primalscheme labs) or a custom primer scheme.
a: ARTIC Primer schemes
ARTIC primer schemes should be provided with the scheme_name field in the samplesheet. The example in the guide below uses the artic-measles/400/v1.0.0 scheme, however this will change depending on the primer scheme that was used used to generate the data. If you are unsure, check the name on primalscheme labs and ensure it follows the pattern <SCHEME>/<SCHEME_LENGTH>/<VERSION>.
The pipeline will automatically find the scheme and download it for you if you do this.
b: Custom Primer Schemes
If you have used a primer scheme which is not on the ARTIC primerschemes repository, you will have to provide two different fields in the samplesheet, custom_scheme_path and custom_scheme_name.
Your custom scheme files must be named like this:
/some/directory/custom_scheme
├── primer.bed
└── reference.fasta
In which case you would provide the custom_scheme_path of /some/directory/custom_scheme and a custom_scheme_name which reflects your custom scheme in the samplesheet.
2) Oxford Nanopore (ONT) specific set up
You will need to provide information to amplicon-nf about where the read data is located and what the barcodes included in your sequencing run are.
It is important to remember that for ONT data the pipeline expects a directory of FASTQ files to be provided, not fastq files directly even if a directory only contains a single FASTQ file.
If you are running ONT sequenced samples through the pipeline, the fastq_pass directory will likely have a file structure like this:
/some/directory/fastq_pass
├── barcode01
│ ├── reads0.fastq.gz
│ └── reads1.fastq.gz
├── barcode02
│ ├── reads0.fastq.gz
│ ├── reads1.fastq.gz
│ └── reads2.fastq.gz
└── barcode03
└── reads0.fastq.gz
There are two ways to provide the location of the sequencing read data for ONT data:
a) With implicit (fuzzy) matching of FASTQ directories based on the provided barcode column, this option will match up subdirectories of the directory provided with the --read_directory parameter so you will not have to provide filepaths within the samplesheet.
b) With explicit FASTQ directories within the samplesheet in the fastq_directory column. This approach may reduce the risk of mismatching metadata and read data, however you will need to be able to identify the absolute path to the read directory.
a: Implicit (fuzzy) FASTQ Directory Input
If you wish to utilise fuzzy directory matching then a valid samplesheet could look like this (remember, the fastq_pass directory MUST be provided with read_directory parameter for this samplesheet to be valid):
sample,platform,scheme_name,barcode
sample1,nanopore,artic-measles/400/v1.0.0,barcode01
sample2,nanopore,artic-measles/400/v1.0.0,barcode02
sample3,nanopore,artic-measles/400/v1.0.0,barcode03
Please make sure that you provide a barcode which matches the directory exactly, for example,
01would be invalid since the actual directory isbarcode01, if thebarcodecolumn does not match the directory EXACTLY then the pipeline will not be able to match the FASTQ files with the metadata.
We have generated an example implicit Nanopore samplesheet google sheet which is available here for your reference.
You may wish to copy this and use it as a basis for your own samplesheets, to download a samplesheet CSV which is compatible with the pipeline; press “File”, “Download”, then “Comma Separated Values (.csv)”.
b: Explicit FASTQ Directory Input
If you wish to provide explicit FASTQ directories then a valid samplesheet could look like this:
sample,platform,scheme_name,fastq_directory
sample1,nanopore,artic-measles/400/v1.0.0,/some/directory/fastq_pass/barcode01
sample2,nanopore,artic-measles/400/v1.0.0,/some/directory/fastq_pass/barcode02
sample3,nanopore,artic-measles/400/v1.0.0,/some/directory/fastq_pass/barcode03
An example explicit Nanopore samplesheet google sheet which is available here for your reference.
You may wish to copy this and use it as a basis for your own samplesheets, to download a samplesheet CSV which is compatible with the pipeline; press “File”, “Download”, then “Comma Separated Values (.csv)”.
3) Illumina specific set up
If you are running Illumina sequenced samples through the pipeline then you only need to fill in a subset of the samplesheet. The setup instructions assume the Illumina data directory looks like this:
/some/directory/run_fastq_directory
├── sample-1_S1_R1_001.fastq.gz
│── sample-1_S1_R2_001.fastq.gz
├── sample-2_S2_R1_001.fastq.gz
└── sample-2_S2_R2_001.fastq.gz
As with ONT data, there are two ways to setup samplesheets for Illumina datasets;
a) With implicit (fuzzy) matching of FASTQ file pairs based on the provided sample column, this option will match up file pairs within the directory provided with the --read_directory parameter so you will not have to provide filepaths within the samplesheet.
b) With explicit FASTQ directories within the samplesheet in the fastq_1 and fastq_2 columns. This approach may reduce the risk of mismatching metadata and read data, however you will need to be able to identify the absolute path to the read directory.
a: Implicit (fuzzy) matched paired FASTQ Input
If you wish to utilise fuzzy directory matching then a valid samplesheet could look like this (remember, the run_fastq_directory path MUST be provided with the read_directory parameter for this samplesheet to be valid).
sample,platform,scheme_name
sample-1,illumina,artic-measles/400/v1.0.0
sample-2,illumina,artic-measles/400/v1.0.0
We have generated an example implicit matched Illumina samplesheet which is available here for your reference.
You may wish to copy this and use it as a basis for your own samplesheets, to download a samplesheet CSV which is compatible with the pipeline; press “File”, “Download”, then “Comma Separated Values (.csv)”.
More information on samplesheet generation is included in the amplicon-nf repository including information about how to utilise custom schemes and more, the information in that document is primarily for command line users but the descriptions of how to properly format your samplesheet CSV is applicable here too.
b: Explicit paired FASTQ input
For explicit FASTQ pairs, a valid samplesheet for the above directory would look like this:
sample,platform,scheme_name,fastq_1,fastq_2
sample-1,illumina,artic-measles/400/v1.0.0,/some/directory/run_fastq_directory/sample-1_S1_R1_001.fastq.gz,/some/directory/run_fastq_directory/sample-1_S1_R2_001.fastq.gz
sample-2,illumina,artic-measles/400/v1.0.0,/some/directory/run_fastq_directory/sample-2_S2_R1_001.fastq.gz,/some/directory/run_fastq_directory/sample-2_S2_R2_001.fastq.gz
We have generated an example explicit Illumina samplesheet google sheet which is available here for your reference.
You may wish to copy this and use it as a basis for your own samplesheets, to download a samplesheet CSV which is compatible with the pipeline; press “File”, “Download”, then “Comma Separated Values (.csv)”.
Running amplicon-nf using EPI2ME Desktop
1) Starting EPI2MElabs Desktop and Installing amplicon-nf
Now you have a samplesheet prepared for your data you have everything you need to begin processing it with amplicon-nf, first open the EPI2ME desktop client, when you do you should see a window which looks like this:
If you have a Nanopore account you may wish to sign in, however, if you do not wish to there is a hidden menu on the ... next to “Oxford Nanopore Technologies 2025” which will reveal a hidden “Continue as guest” button, we apologise for how hidden this functionality is, we have no affiliation with epi2me so cannot control their interface.
Once you have logged in or signed in as a guest you will see a page which looks like the screenshot below, on the left side of the window, press the “Launch” button which will take you to the workflows tab.
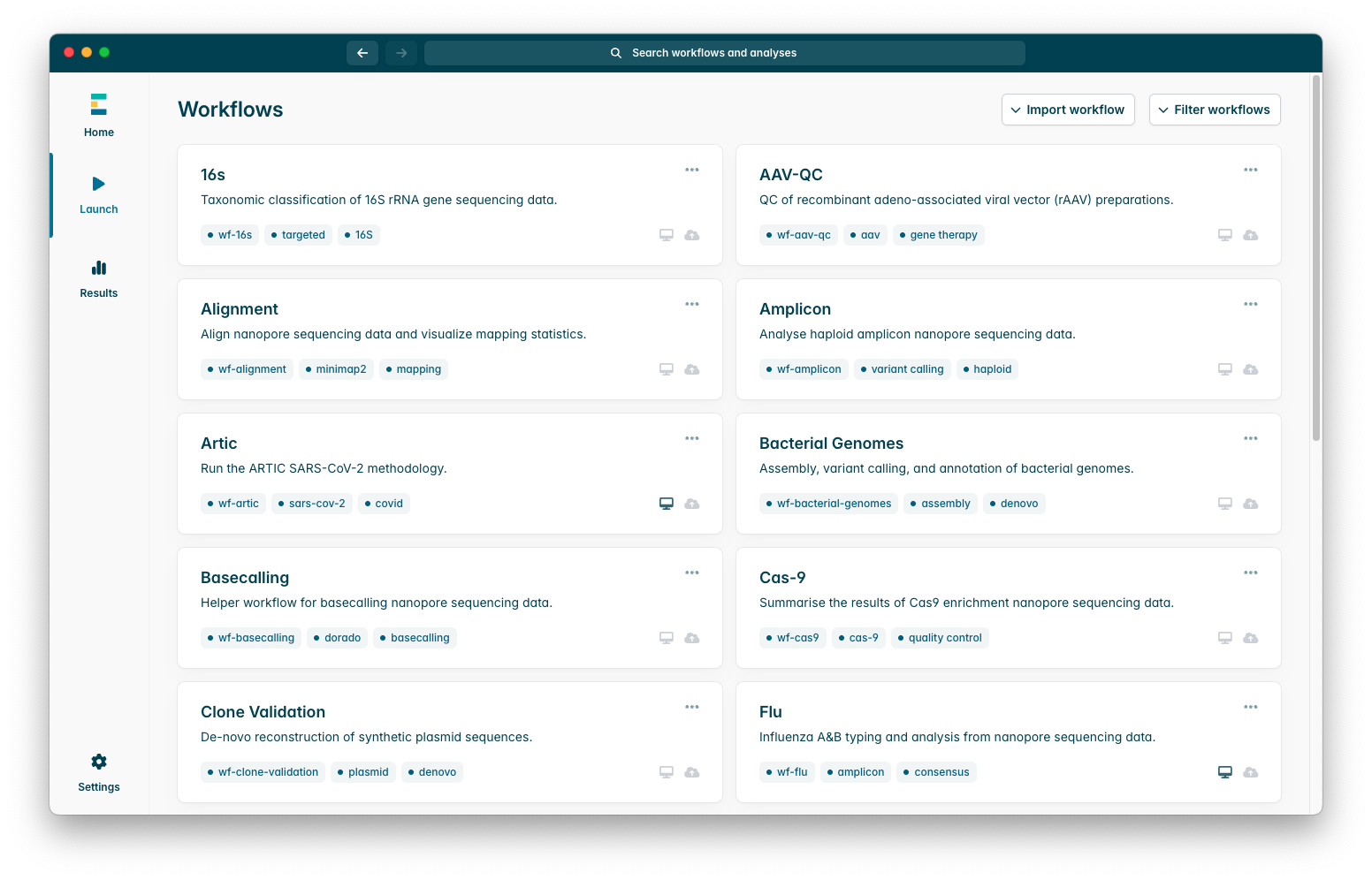
The workflows tab looks like the screenshot below; the pipelines available by default are those developed by in-house EPI2ME developers, amplicon-nf is not installed by default so we will have to import it, to do so press the “Import Workflow” button at the top right of the workflows tab.
You will see two options, “Import from Github” or “Import a 2ME file”, we will be importing the workflow from Github so select that option, you will see a text box which looks like this:
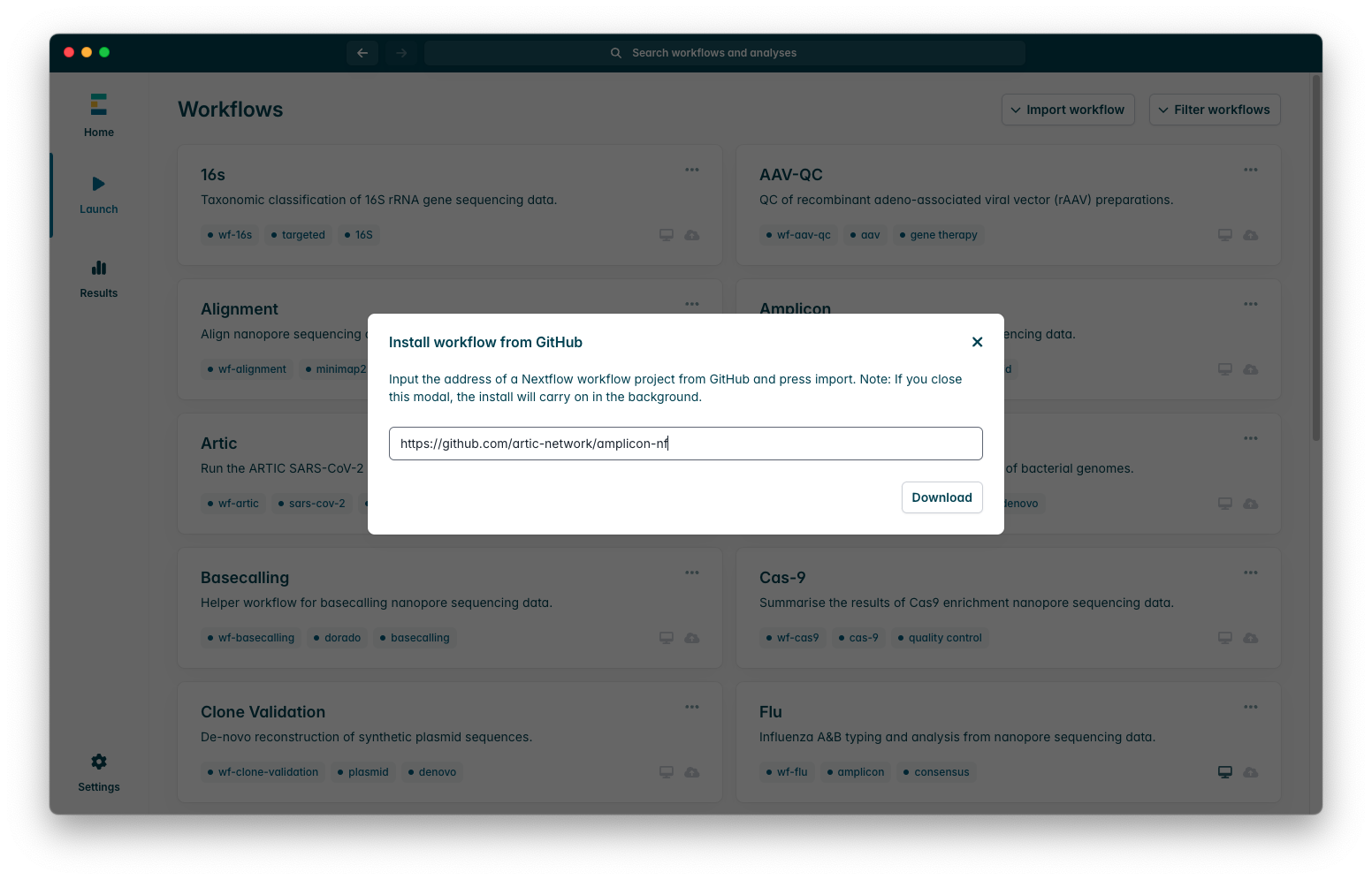
When you do, you will need to enter the amplicon-nf github repository URL, “https://github.com/artic-network/amplicon-nf”, please ensure that you enter the URL exactly as provided here, any differences will mean that the workflow cannot be imported, even leaving a / at the end of the URL can prevent workflows from being imported. Once you have entered the URL as in the screenshot, press “Download”, this download may sometimes fail due to connection errors, if it fails the first time please try again, if this fails consistently please contact EPI2ME for assistance with their client.
Once the workflow has downloaded successfully, you should see a success message as in this screenshot:
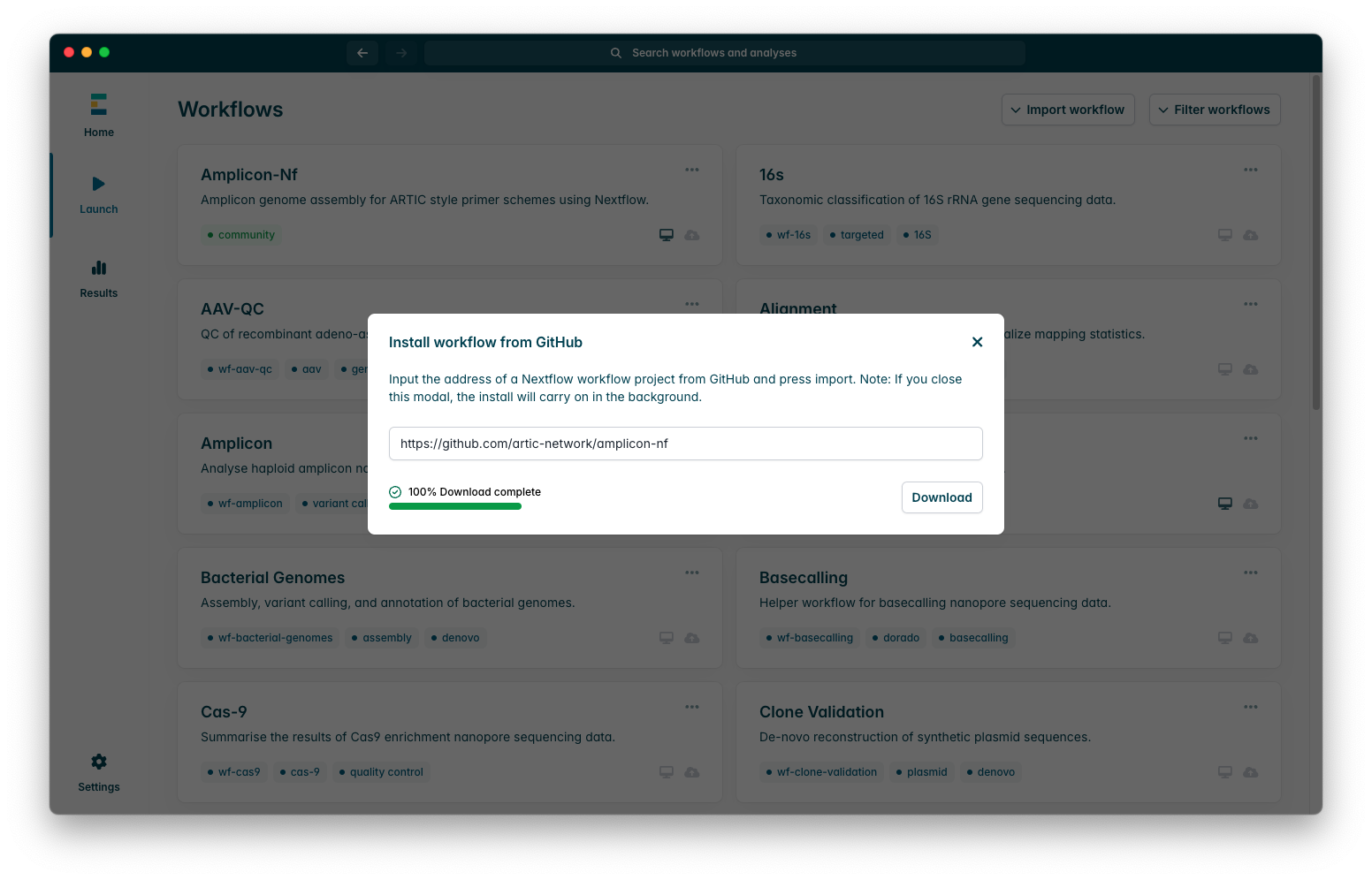
2) Running amplicon-nf
Now you have successfully installed the workflow, you should see it in your workflows tab near the top, as in this screenshot:
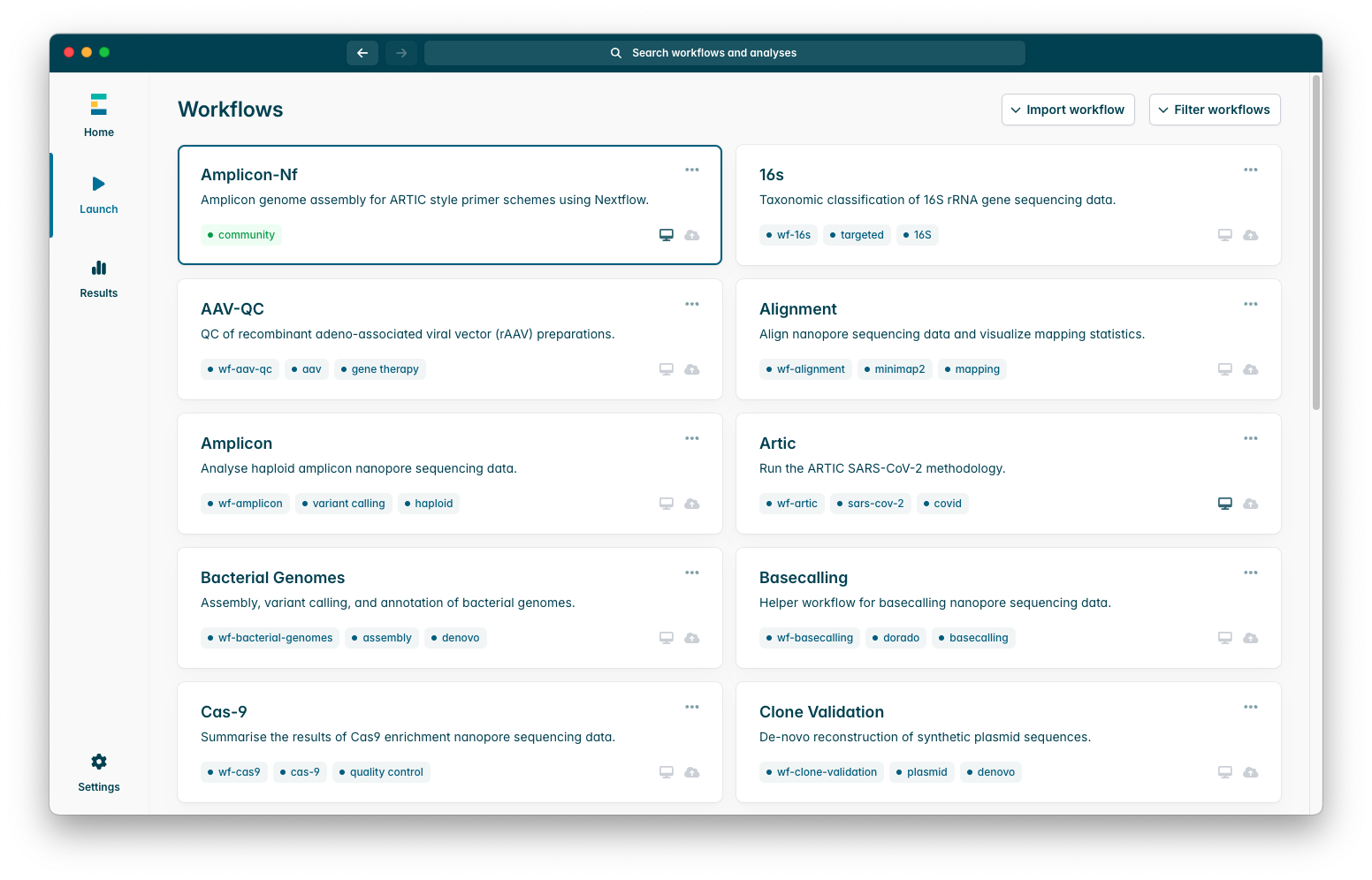
To begin configuring the pipeline, press it, you will see an options menu which looks like this:
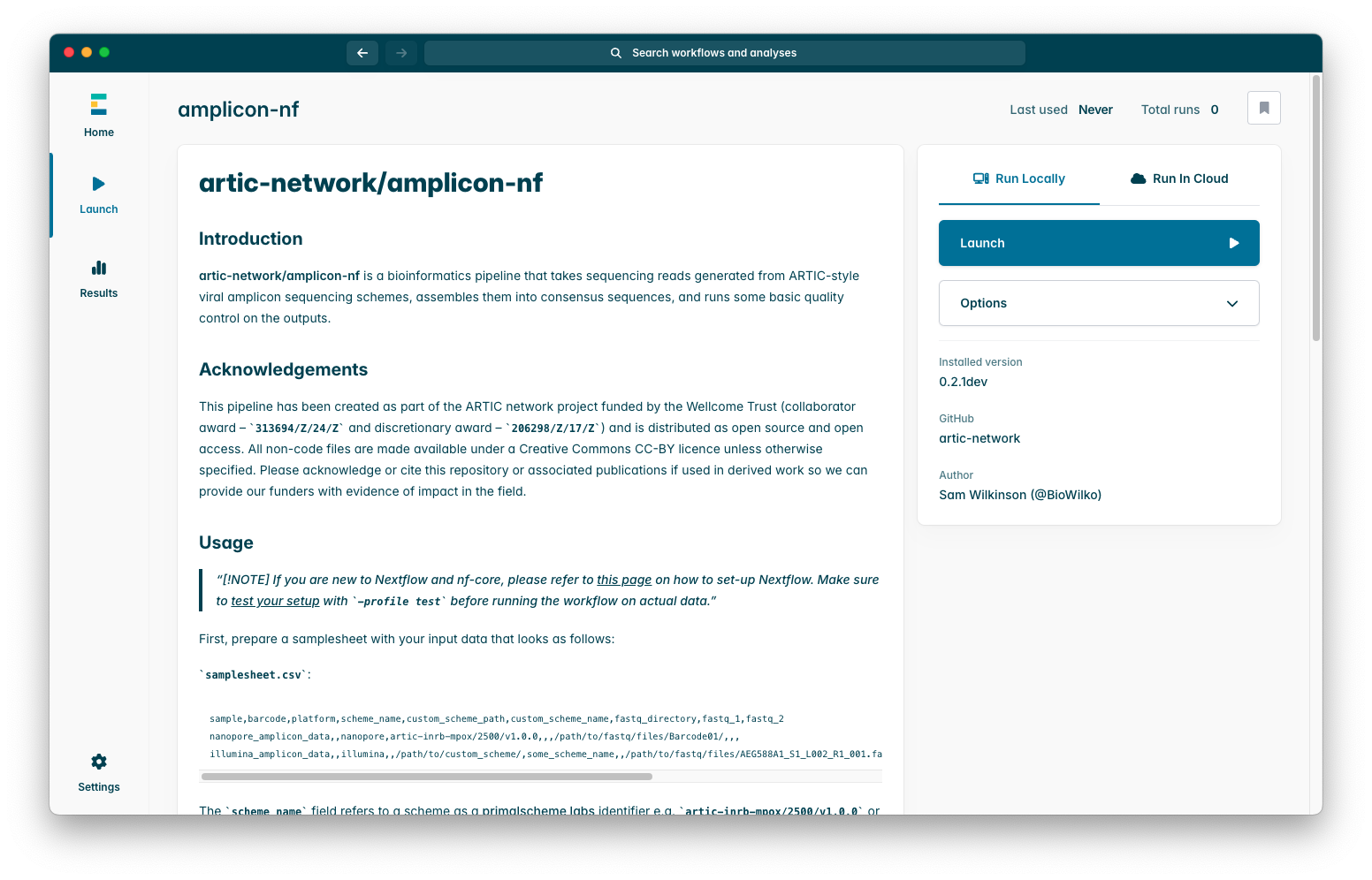
If you had downloaded the pipeline previously it is often a good idea to check for updates, you can do so by pressing “Options”, then “Check for updates”, if you have only just downloaded the pipeline then you can go immediately to the next step which is to press “Launch”, you will see a screen like this:
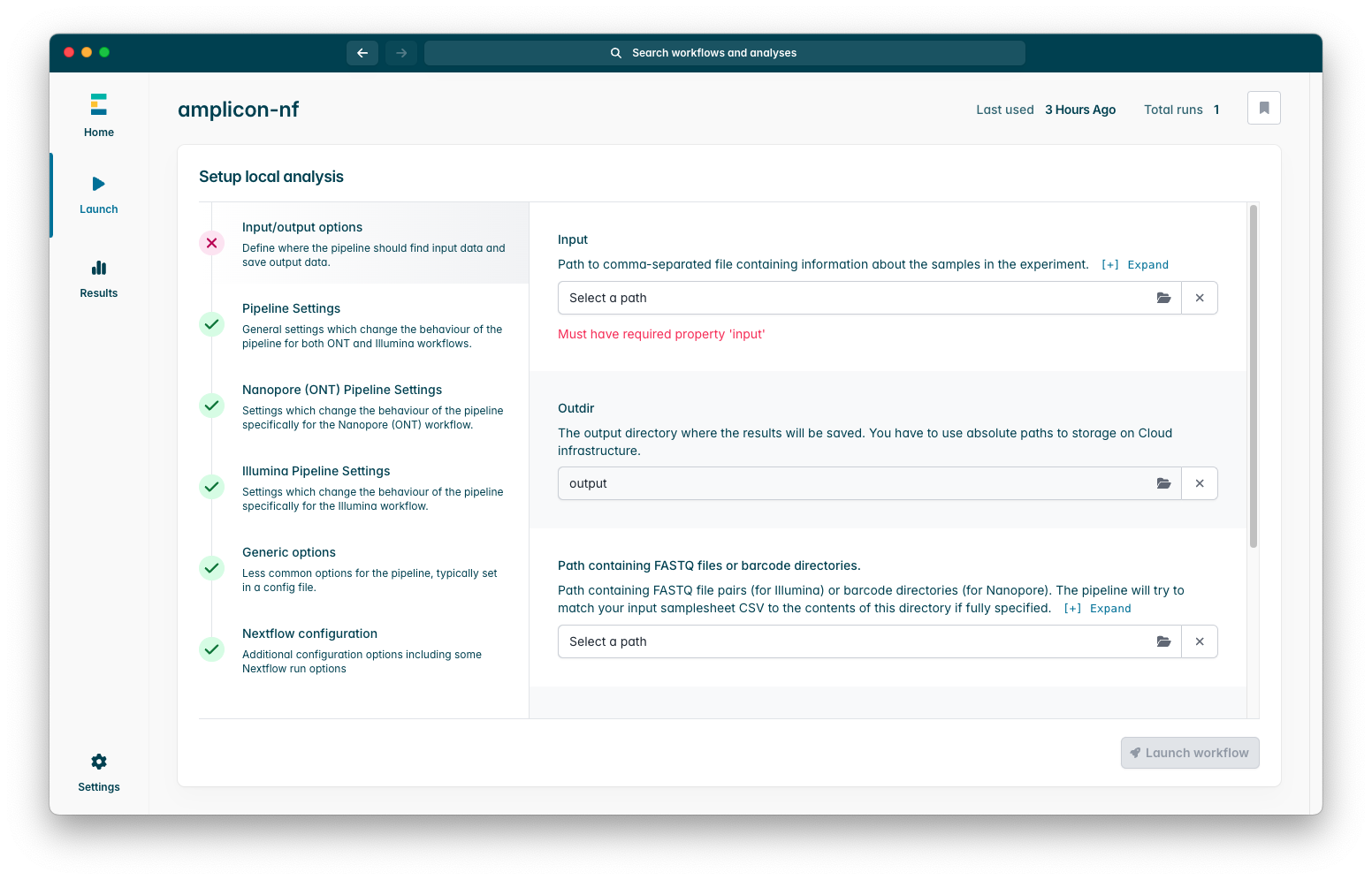
The only strictly required parameter is “Input” which is the run samplesheet we created earlier, if you press on the box which says “Select a path” a window will appear where you can select your samplesheet CSV using your operating systems file browser.
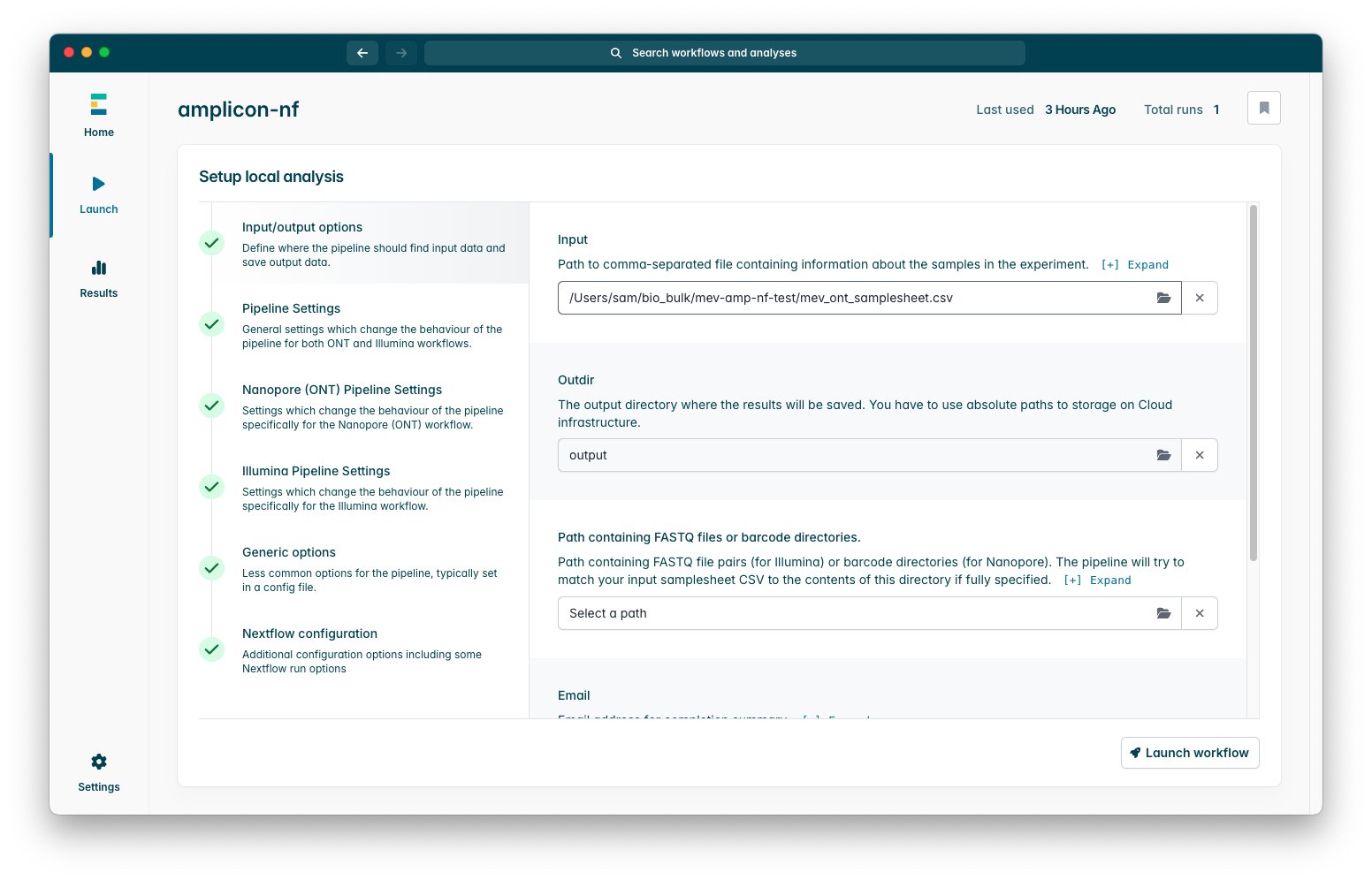
Once you have done so you should see your samplesheet filled into the box, the pipeline will launch now but I will briefly summarise some parameters you may wish to modify:
Input / Output Options
Outdir-> This controls where the pipeline will place output files, if you leave this as the default value it will place this within the epi2melabs directory, we will discuss accessing outputs later.Path containing FASTQ files or directories-> This is required if you have prepared a samplesheet which will utilise the implicit file input matching workflow, this should be the directory that contains your barcode directories or FASTQ file pairs.
Pipeline Settings
Normalise Depth-> This controls to what depth the pipeline will try to normalise your data to before running variant calling, if you set this to 0 the pipeline will not run normalisation at all which will increase your runtime significantly due to the increased complexity of variant calling.
This interface has summary descriptions of what each parameter does as well as detailed help text which you can see if you press the Expand option under the summary, we strongly recommend you familiarise yourself with these so you may modify them to better suit your specific usecase.
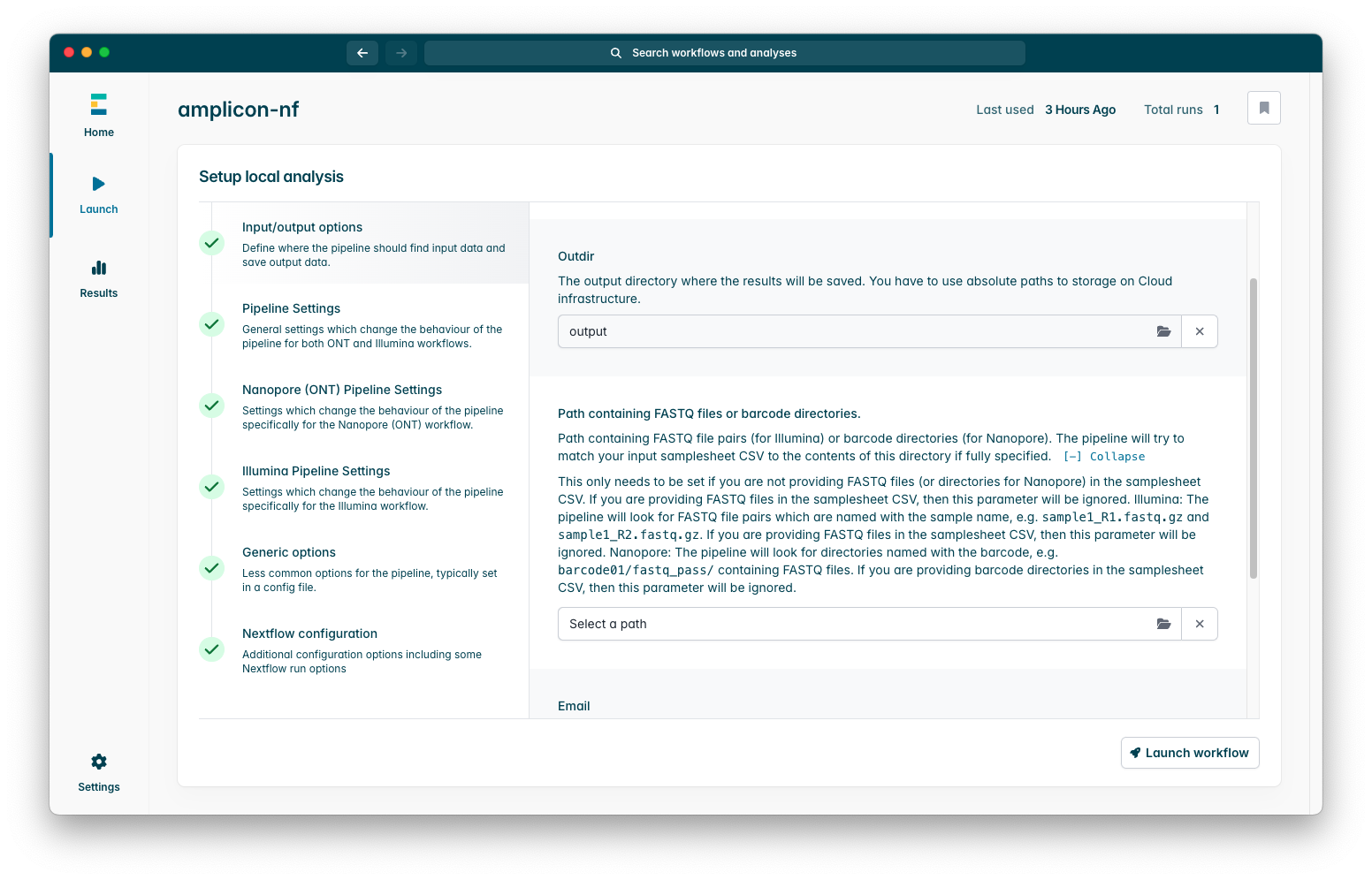
Once you have configured the pipeline as you wish, press the “Launch” button to start the workflow!
3) Workflow Monitoring
Once you launch the workflow you will be shown a run progress screen which will show you currently running processes of the pipeline, this will update as the pipeline runs.
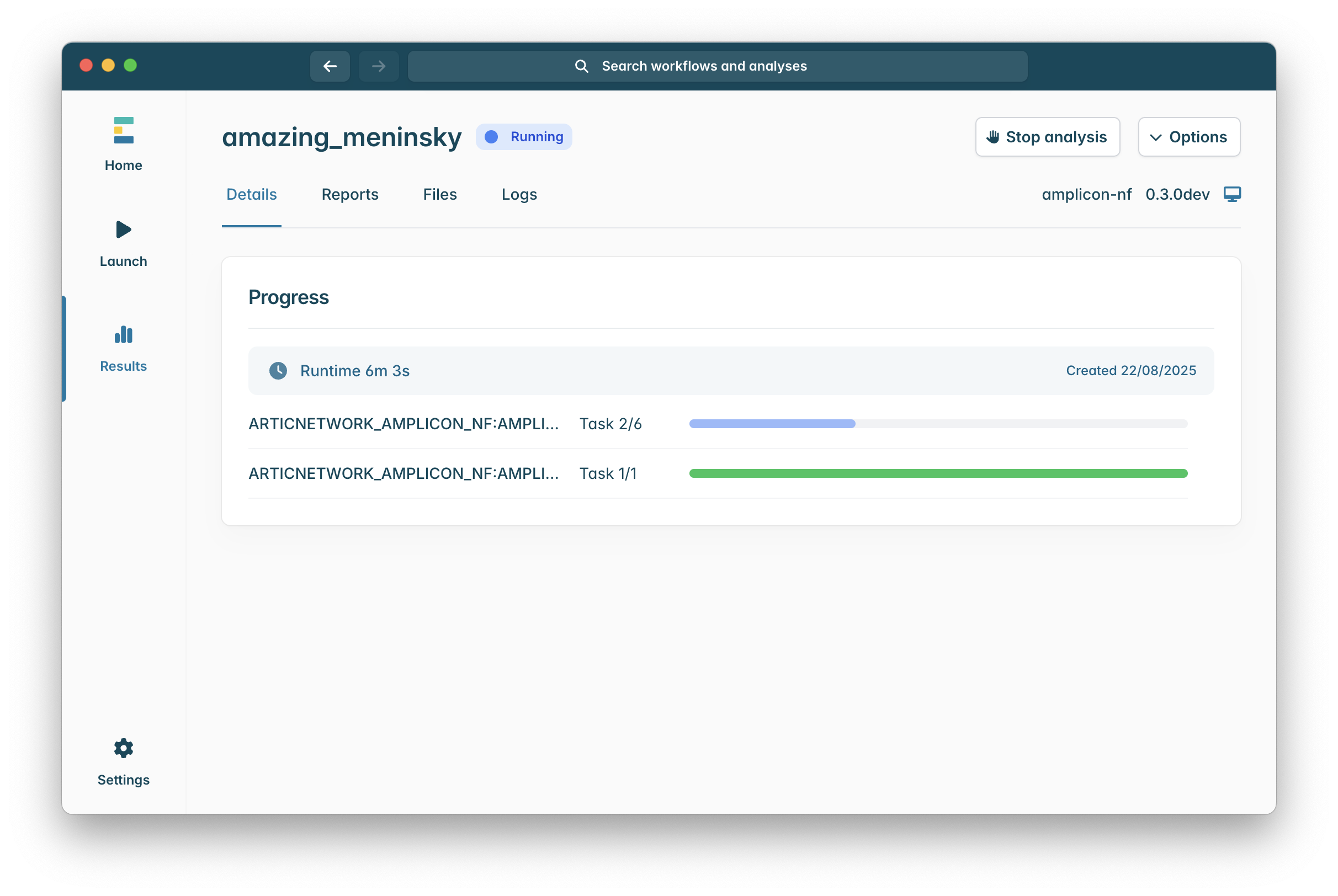
When the workflow has finished the “Running” icon at the top of the window will be replaced with a “Completed icon, sometimes due to how epi2me works a run can be completed some specific processes will not appear to have finished, do not worry if this is the case, so long as you see a green “Completed” as in the image below the pipeline finished successfully!
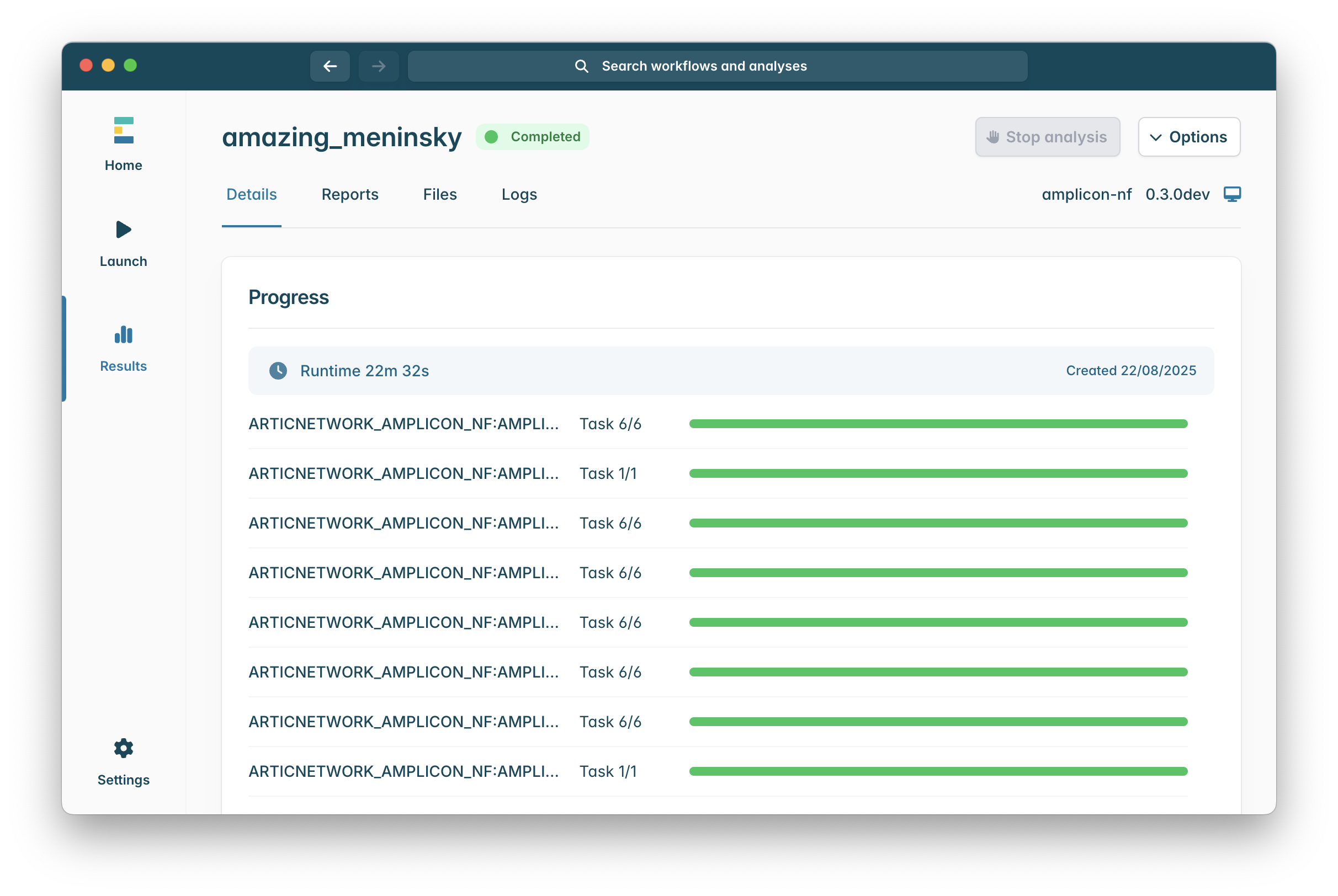
Hopefully you will encounter no issues while running the pipeline, in which case, you might wish to read our epi2me amplicon-nf outputs guide which will walk you through the different output files, their formats, and how to access them from the epi2me client.
However, if you do encounter any issues we have a dedicated document which details problems you may encounter and their possible solutions on the amplicon-nf repository. Please check here when you encounter problems but if your problem is not described there or the solutions do not help please consider creating a github issue so we can discuss it with you directly!
