MPXV alignment and phylogenetics pipeline using Epi2Me
Squirrel | bioinformatics
| Document: | ARTIC-MPXV-phylogeneticsSOP-v1.0 |
| Creation Date: | 2024-08-21 |
| Last Updated: | 2024-08-16 |
| Author: | Áine O'Toole |
| Licence: | Creative Commons Attribution 4.0 International License |
Rationale
MPXV is a large poxvirus, with a complex dsDNA genome ~200kb in length. Alignment of sequences, and therefore phylogenetics, is challenging using classic due to tracts of low-complexity and repetitive regions. Squirrel provides an efficient map-to-reference alignment pipeline with masking of problematic regions of the genome.
Command line
Squirrel can be used as a command line tool, with full command-line documentation available on the squirrel GitHub repository at (github.com/aineniamh/squirrel)[https://github.com/aineniamh/squirrel].
User interface
Squirrel can also be run through the EPI2ME user interface. Please first install the EPI2ME desktop application using the provided link. You can then go to ‘available workflows’ then ‘Import workflow’ from https://github.com/artic-network/squirrel-nf as shown below:
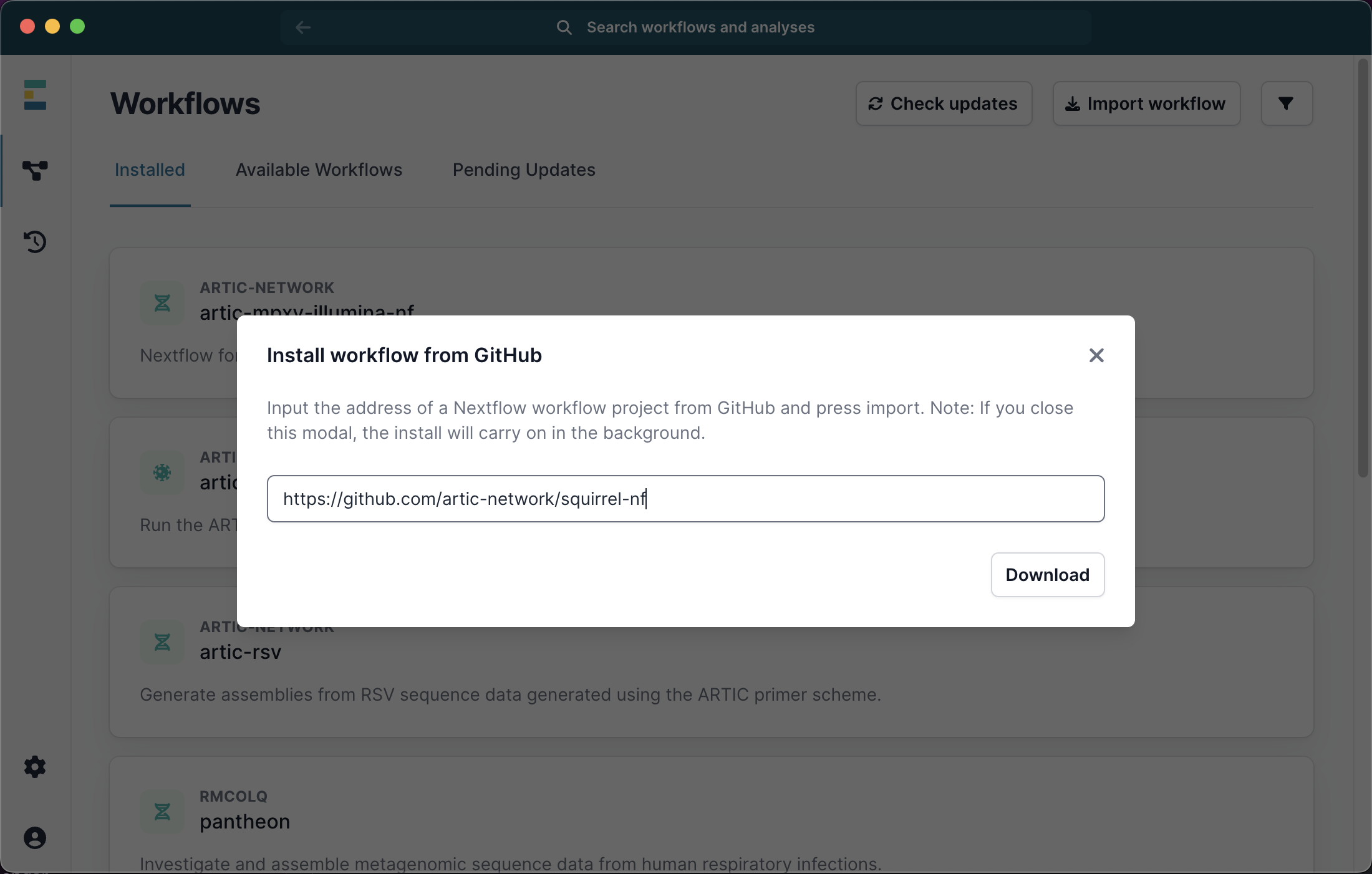
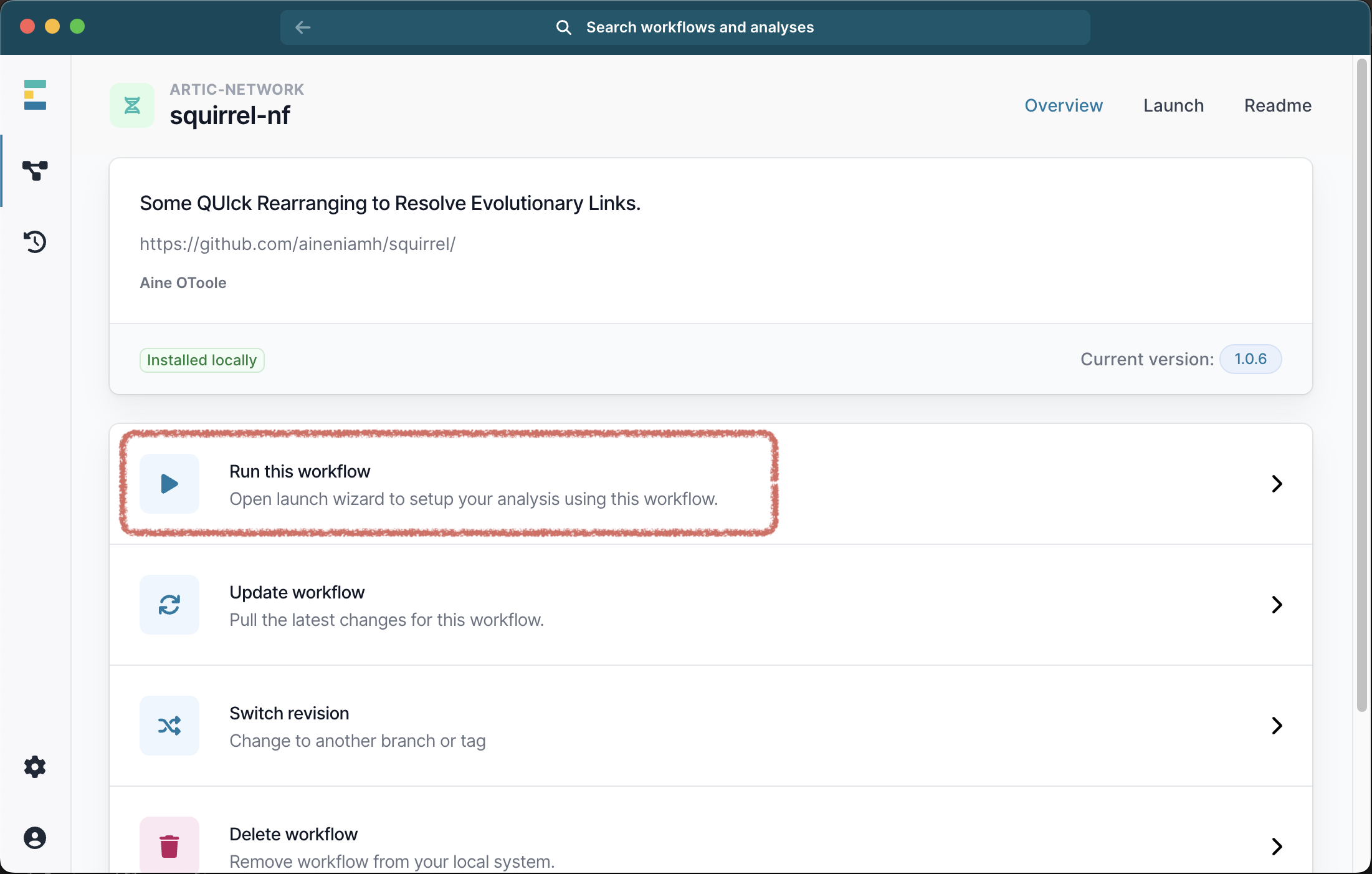
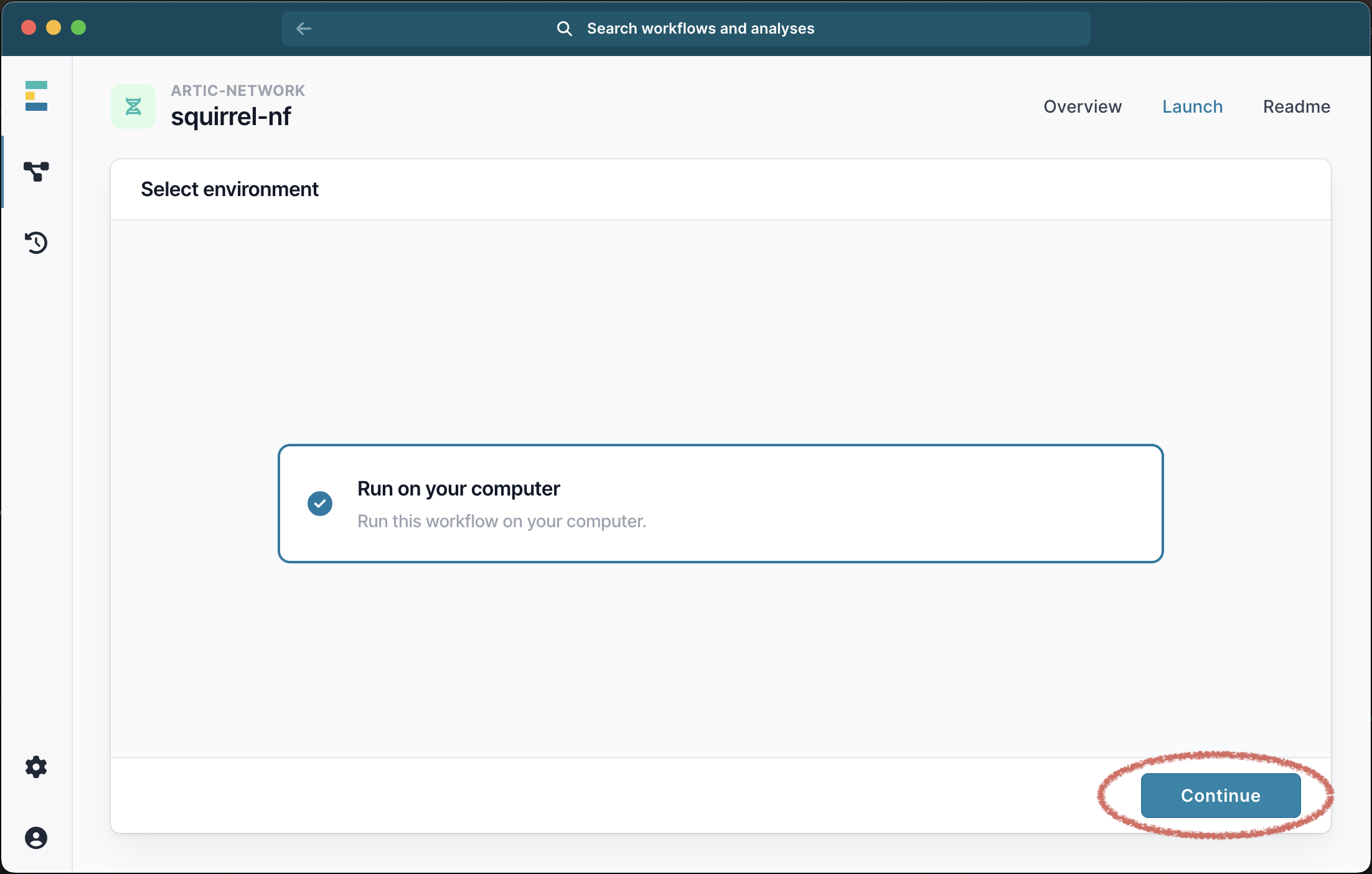
Once the workflow has successfully downloaded, you can click the X to exit to download window, and select it from the list of available workflows. Next select Run this workflow from the available options, and then Run on your computer:
This will bring up a menu where you can provide the inputs for your analysis. The only required file is a single FASTA file containing all the sequences and outgroups for your analysis and you must also select the clade (i or ii) from the drop down list:
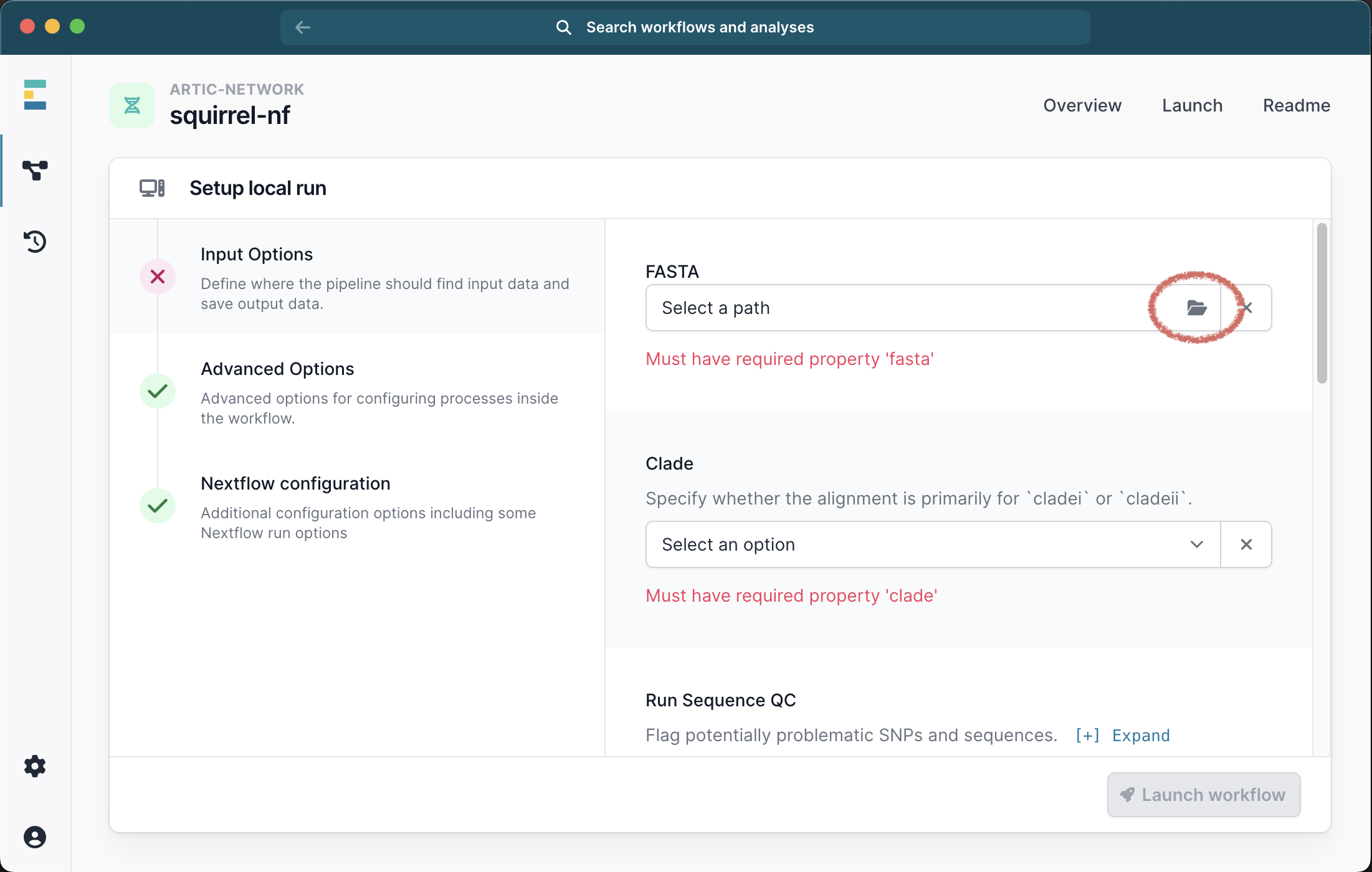
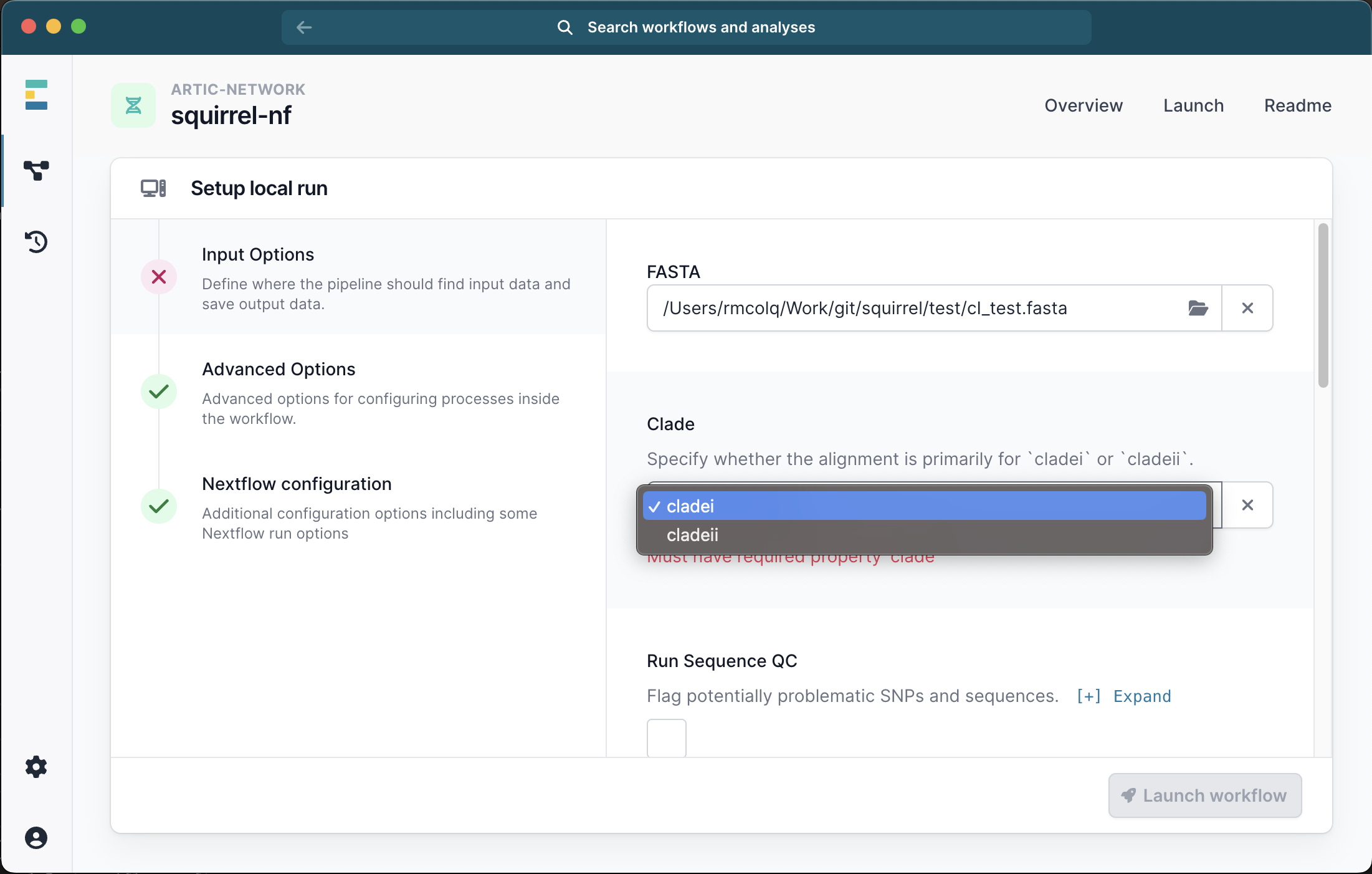
Running with just a FASTA file will generate an alignment of the input sequences. We recommend selecting the check box for Seq QC to check this alignment for problematic sites.
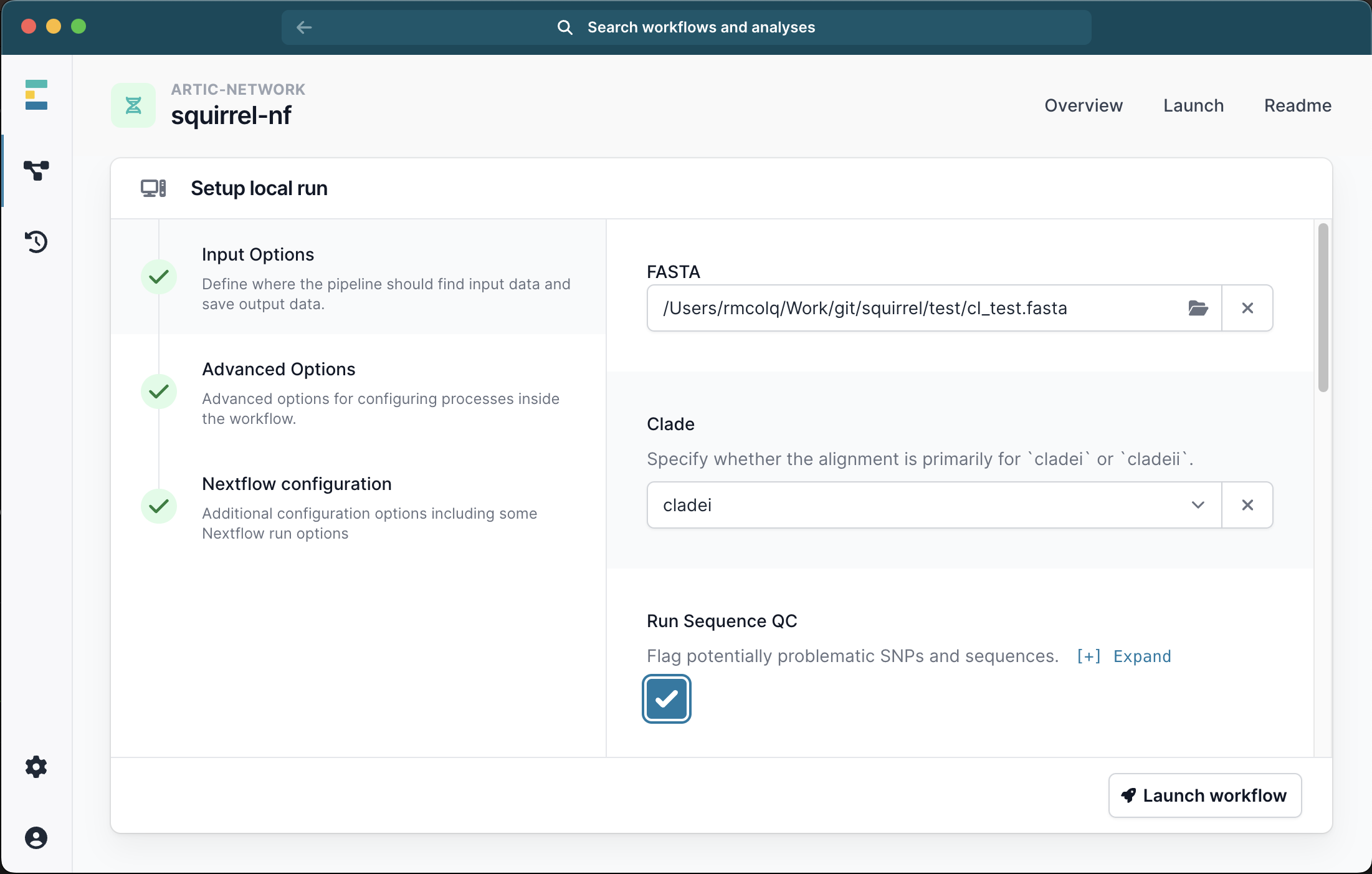
Scrolling down the menu, select the box to Run Phylo. At this point you have 2 options. EITHER you can select the check box to Include Background, in which case a default panel of clade-specific outgroups sequences will be used.
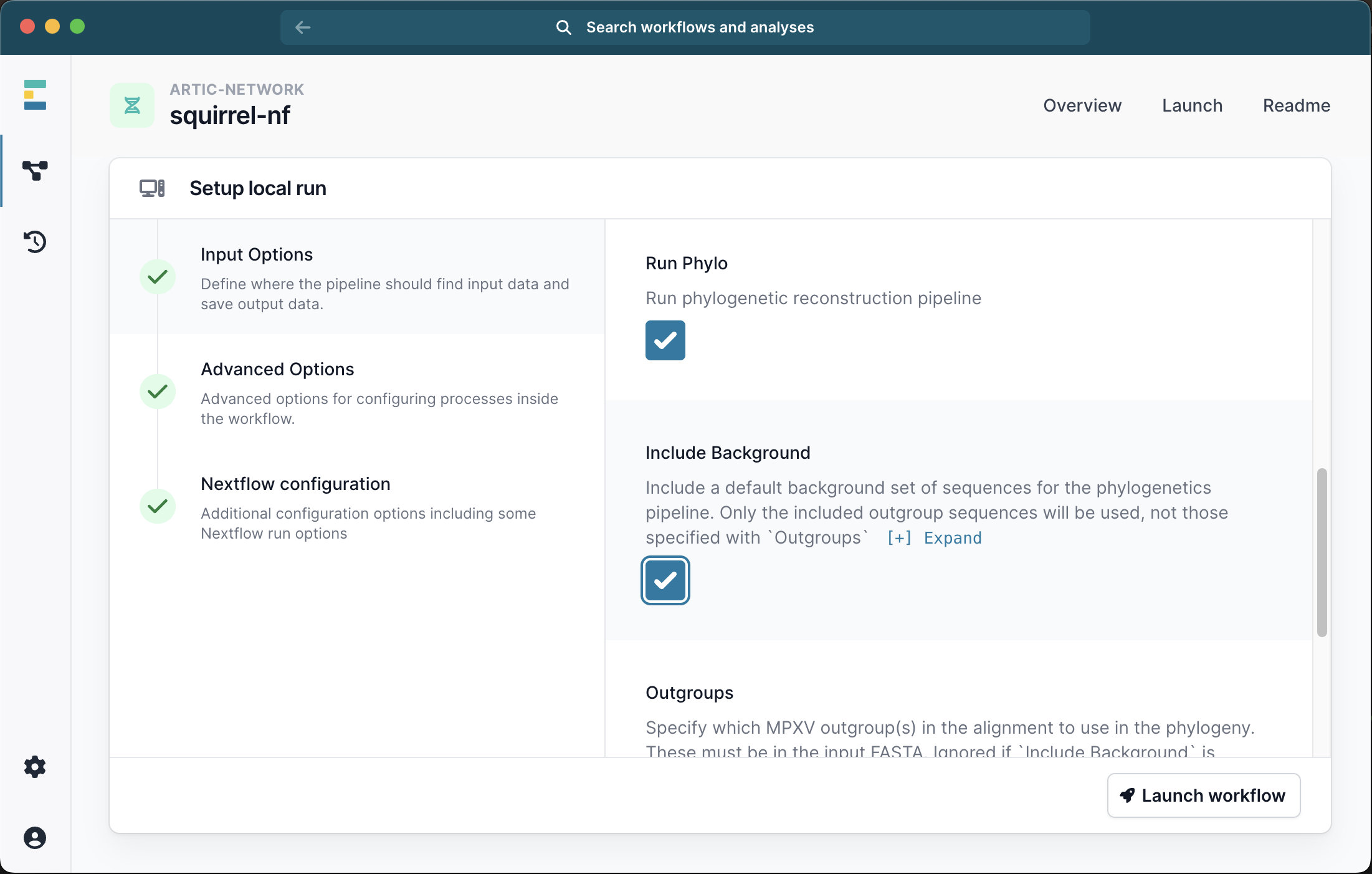
OR you can specify a number of outgroups IDs. These outgroups must be present in the FASTA file you provided and will be pruned out of the final alignment. For Clade I we recommend outgroups KJ642617,KJ642615,KJ642616 and for Clade IIb we recommend KJ642617,KJ642615. If you also selected the Include Background option your specified outgroups will be ignored.
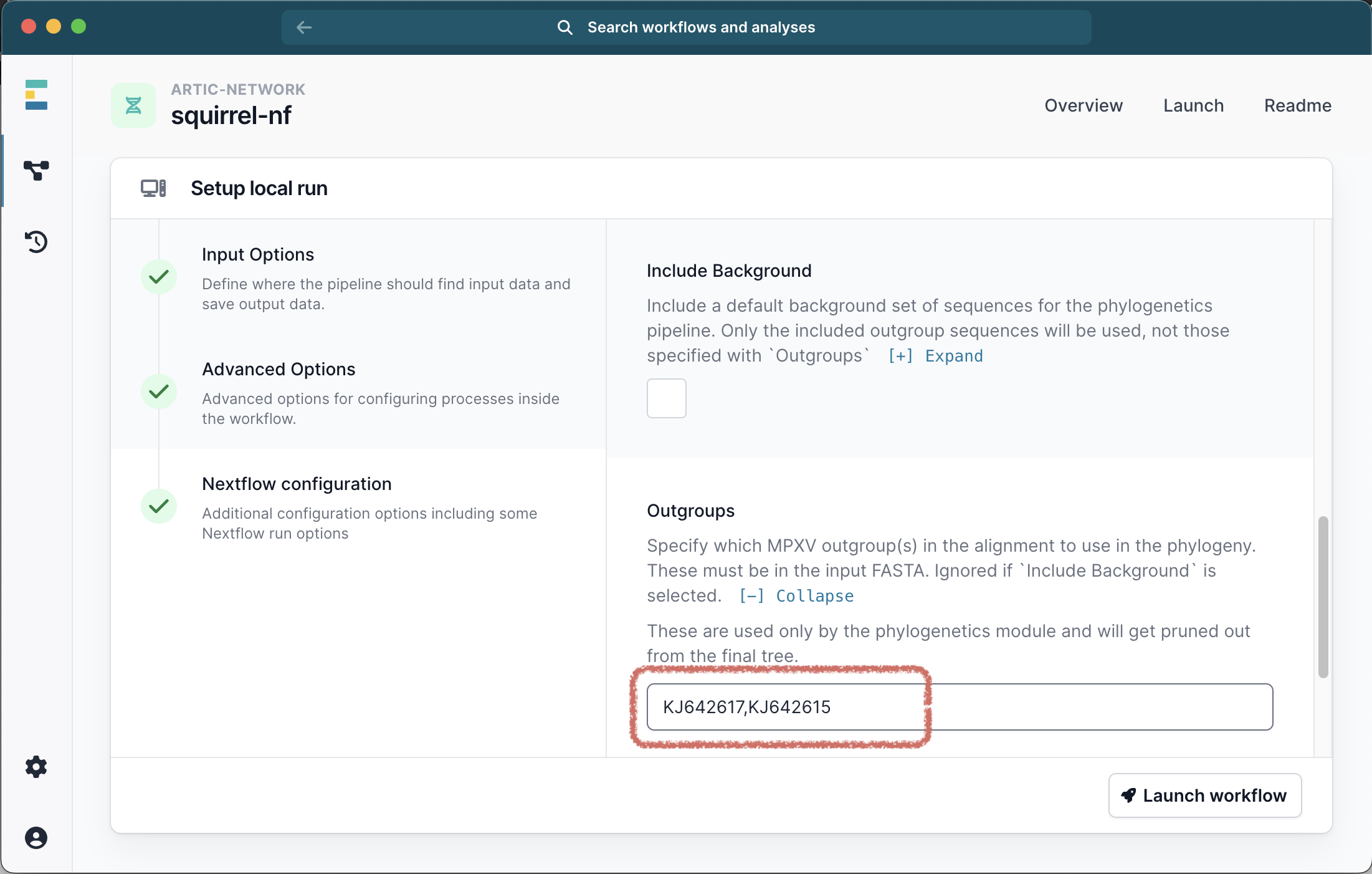
Optionally you can provide a different reference sequence, but this is usually unnecessary - a clade specific reference will be used by default. No Advanced Options or Nextflow Configuration options are required by default.
Click Launch:
This will start the workflow. A progress bar is displayed with the run status but you will not be able to see the stdout that is generated on the command line.
Once the run is completed, a number of files will be available and you can double-click to view them:
This includes a suggested_mask.csv file generated by the run with potentially problematic sites. If you start a new run with the same inputs and additionally provide this mask file in the menu, it will improve the alignment and phylogeny.
Related documents:
- This document is part of the MPXV sequencing protocol package:
- http://artic.network/mpxv/
- Setting up the laptop computing environment using Conda:
- http://artic.network/ebov/ebov-it-setup.html
