Phylogenetic Analysis using Raccoon-nf
| Document: | phylogenetic_analysis-v1.0 |
| Creation Date: | 2026-03-30 |
| Last Updated: | 2026-03-30 |
| Author: | Áine O'Toole, Kate Duggan & Daniel Maloney |
| Licence: | Creative Commons Attribution 4.0 International License |
This tutorial is a walkthrough of running raccoon-nf on the data produced in the workshop. For a complete guide and tutorial see:
Installing and Launching Epi2Me
If EPI2ME isn’t installed, follow these instructions:
1. Launch EPI2ME
Once you have successfully installed, launch EPI2ME. To access EPI2ME without creating an account, click on the three dots at the bottom of the window, and click “Continue as guest”:
2. Import raccoon-nf Workflow
When you have successfully launched EPI2ME, you should see the following screen. Click to open the “Launch” window in the panel on the left hand side:
If it is already installed, click on the raccoon-nf workflow:
and then jump to Step 3.
If raccoon-nf workflow isn’t installed then click on “Import workflow” in the top right of the window, and then “Import from GitHub”:
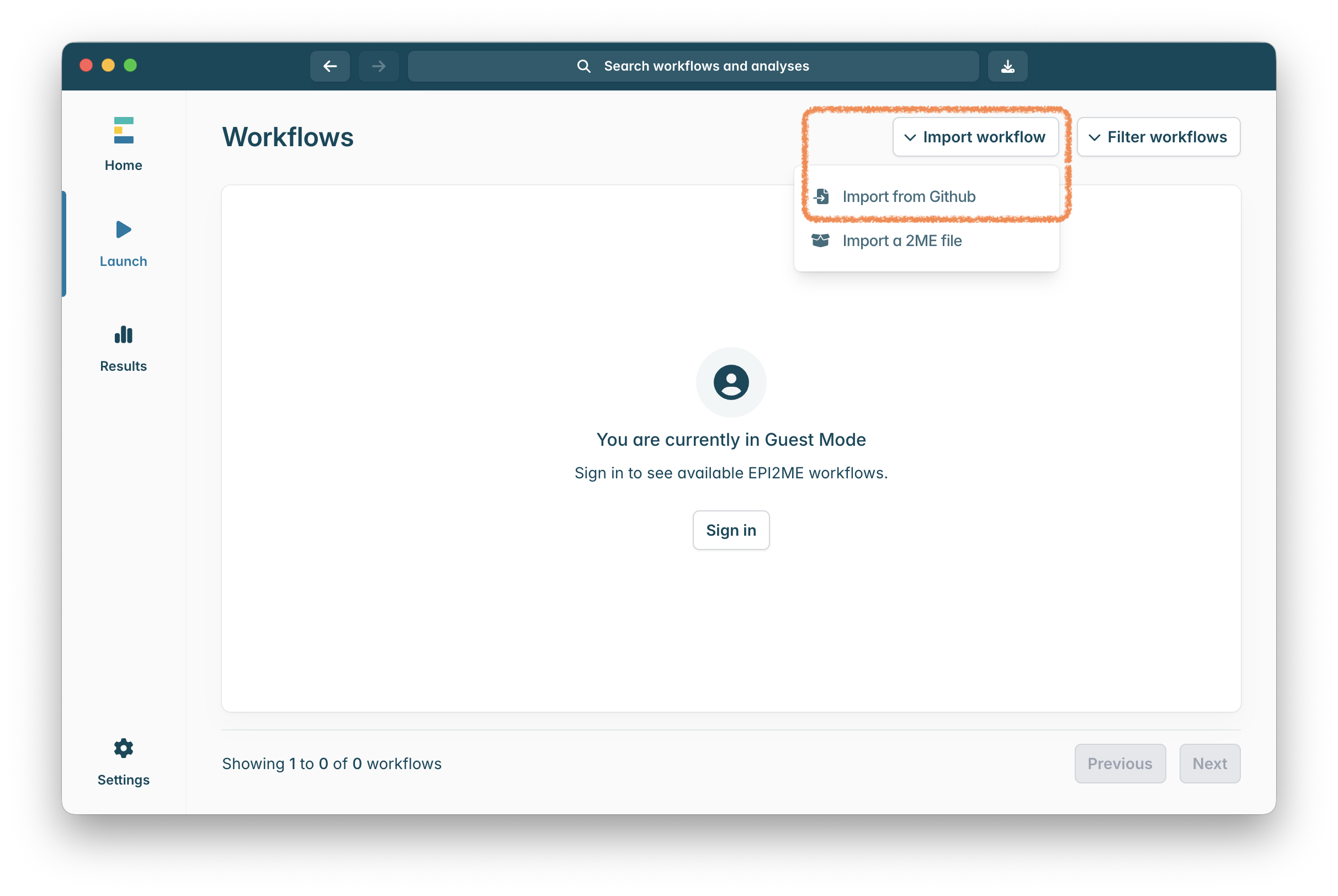
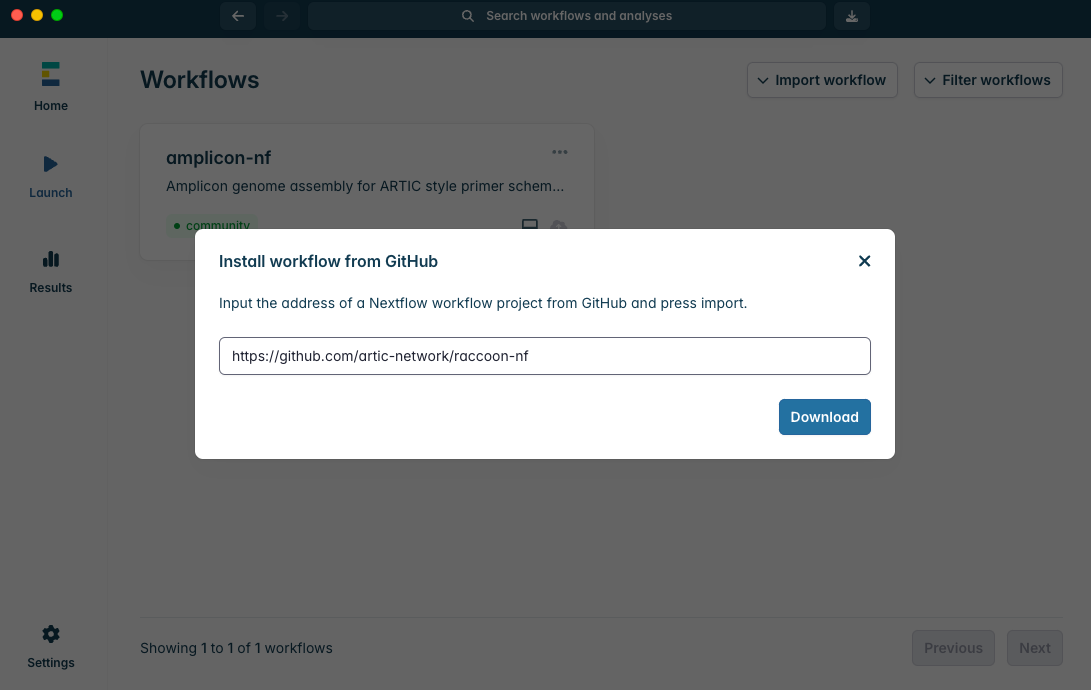
Install amplicon-nf by pasting https://github.com/artic-network/raccoon-nf into the box and clicking “Download”:
3. Setting up the input data for raccoon
To run the raccoon-nf workflow on your data you will need some additional files:
-
A
fastafile containing background genomes from previous outbreaks of LHFV. -
A CSV file containing the metadata for the samples that have been sequenced.
For this workshop these can be downloaded from here:
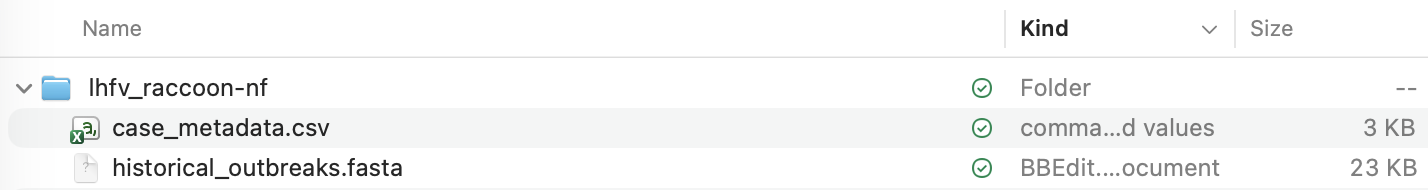
Uncompressing (expanding) this ZIP file will produce a folder with the required files in it:
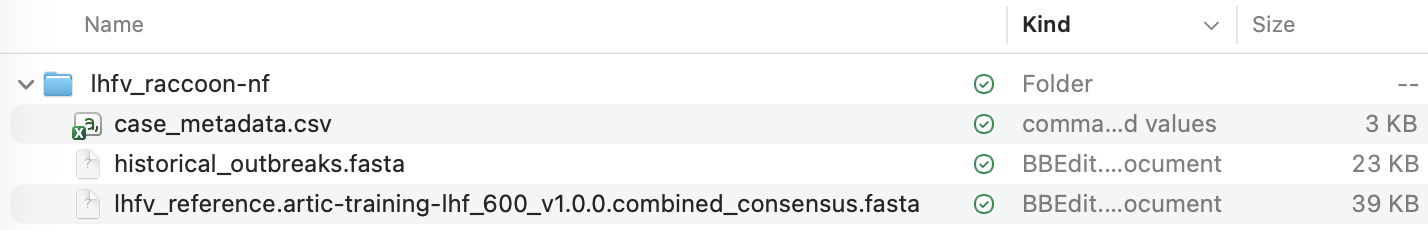
Now copy the consensus genome fasta file from the output of amplicon-nf into this folder. This file will be called lhfv_reference.artic-training-lhf_600_v1.0.0.combined_consensus.fasta:
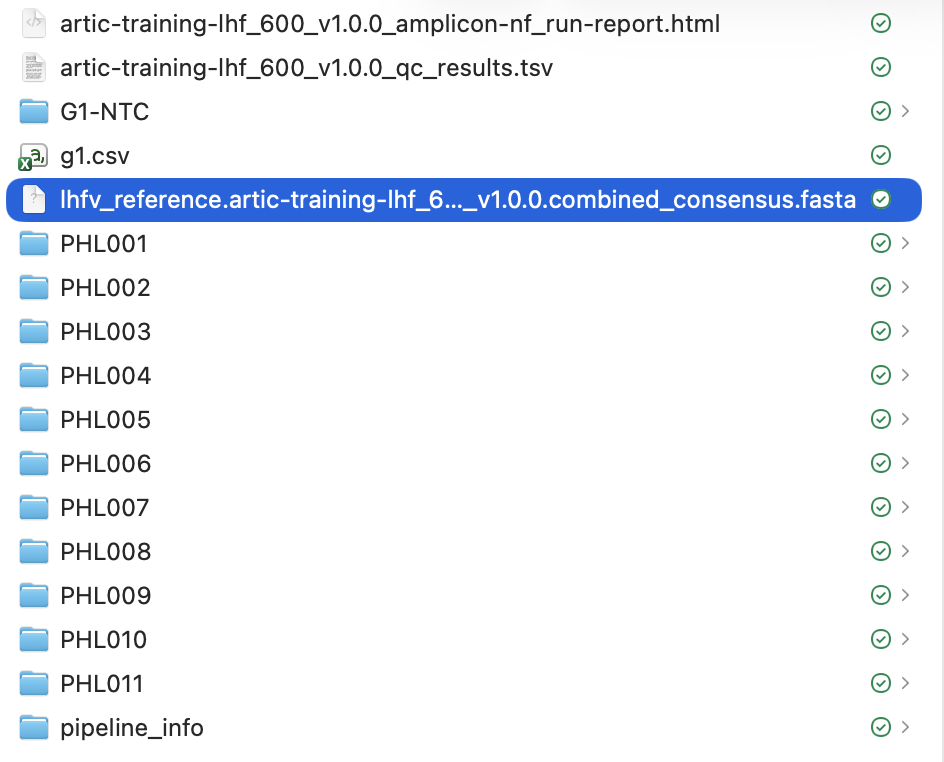
After copying this file the lhfv_raccoon-nf should look like this:
4. Running the raccoon-nf workflow
Click on the ‘raccoon-nf’ workflow to open it:
Click the ‘Launch’ button to start an instance of this workflow.
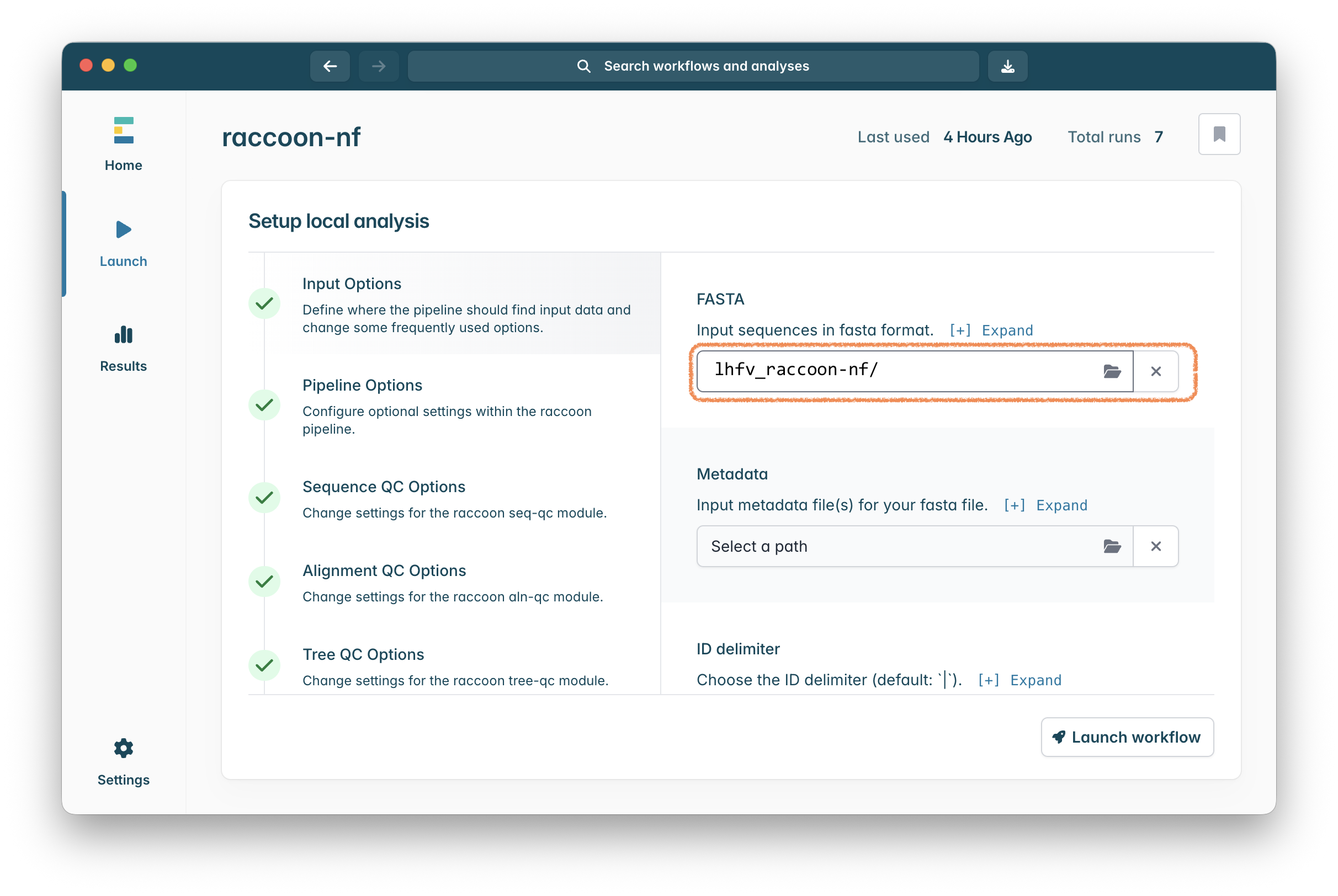
Click on “Select a path” in the “Input sequences in fasta format” setting and in the file chooser that appears select the lhfv_raccoon-nf folder. Raccoon will pick up both the fasta files in here (your data and the background data) and combine them automatically:
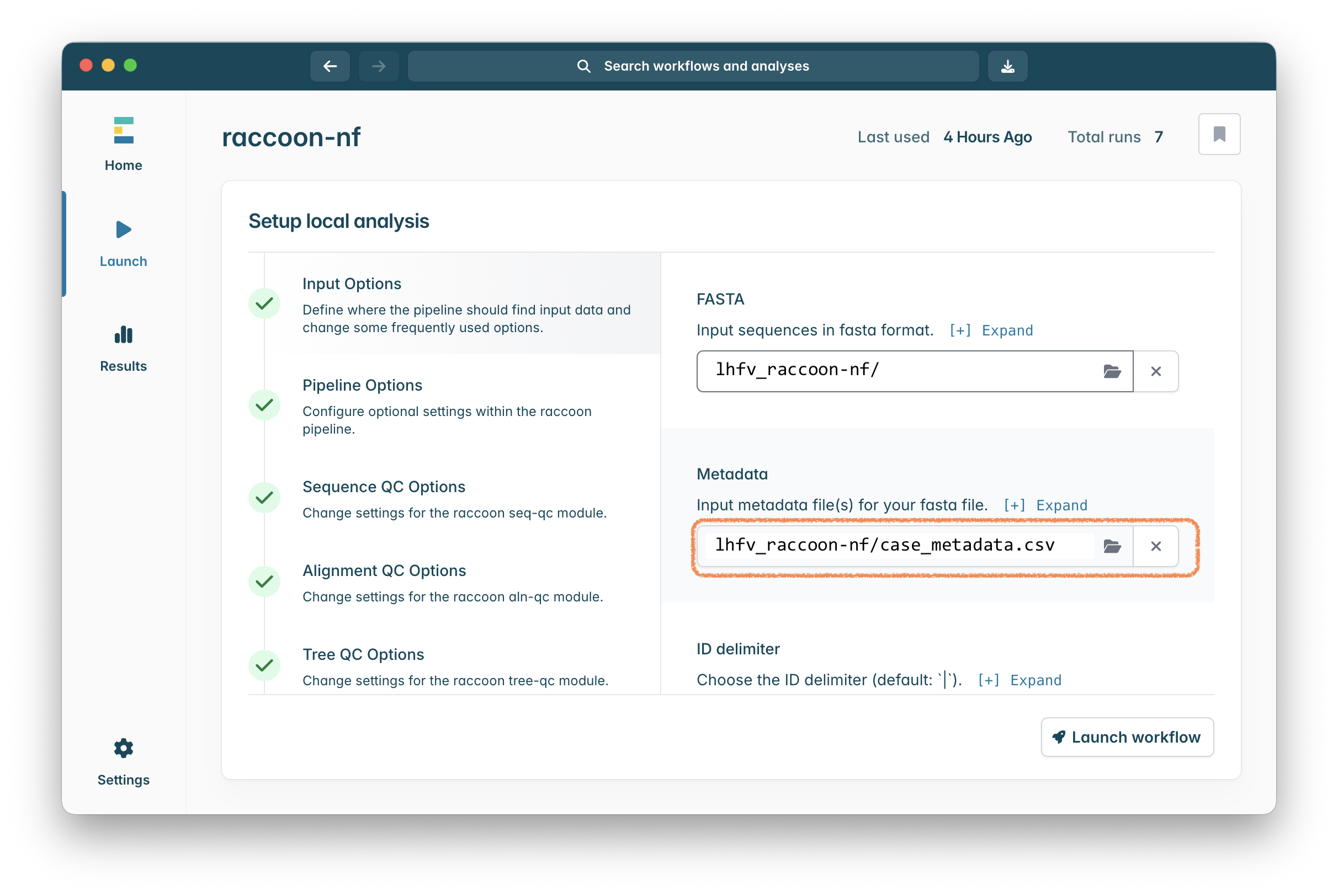
Click on “Select a path” in the “Input metadata file(s) for your fasta file.” setting and in the file chooser that appears select the case_metadata.csv file in the lhfv_raccoon-nf folder.
The case_metadata.csv file looks like this, providing location and collection dates for each sample:
sample country location location2 date
PHL001 Articia Mylona_Marsh Moranga_Moor 2025-12-03
PHL002 Articia Mylona_Marsh Moranga_Moor 2025-12-08
PHL003 Articia Mylona_Marsh Moranga_Moor 2025-12-28
PHL004 Articia Mylona_Marsh Quaye_Quay 2026-01-02
PHL005 Articia Mylona_Marsh Donkor_Dale 2026-01-03
PHL006 Articia Mylona_Marsh Elliville 2026-01-05
PHL007 Articia Mylona_Marsh Bedeburgh 2026-01-10
PHL008 Articia Mylona_Marsh Faux_Kent 2026-01-17
PHL009 Articia Mylona_Marsh Moranga_Moor 2026-01-20
.
.
.
Finally click the “Launch workflow” button and wait until the workflow completes:
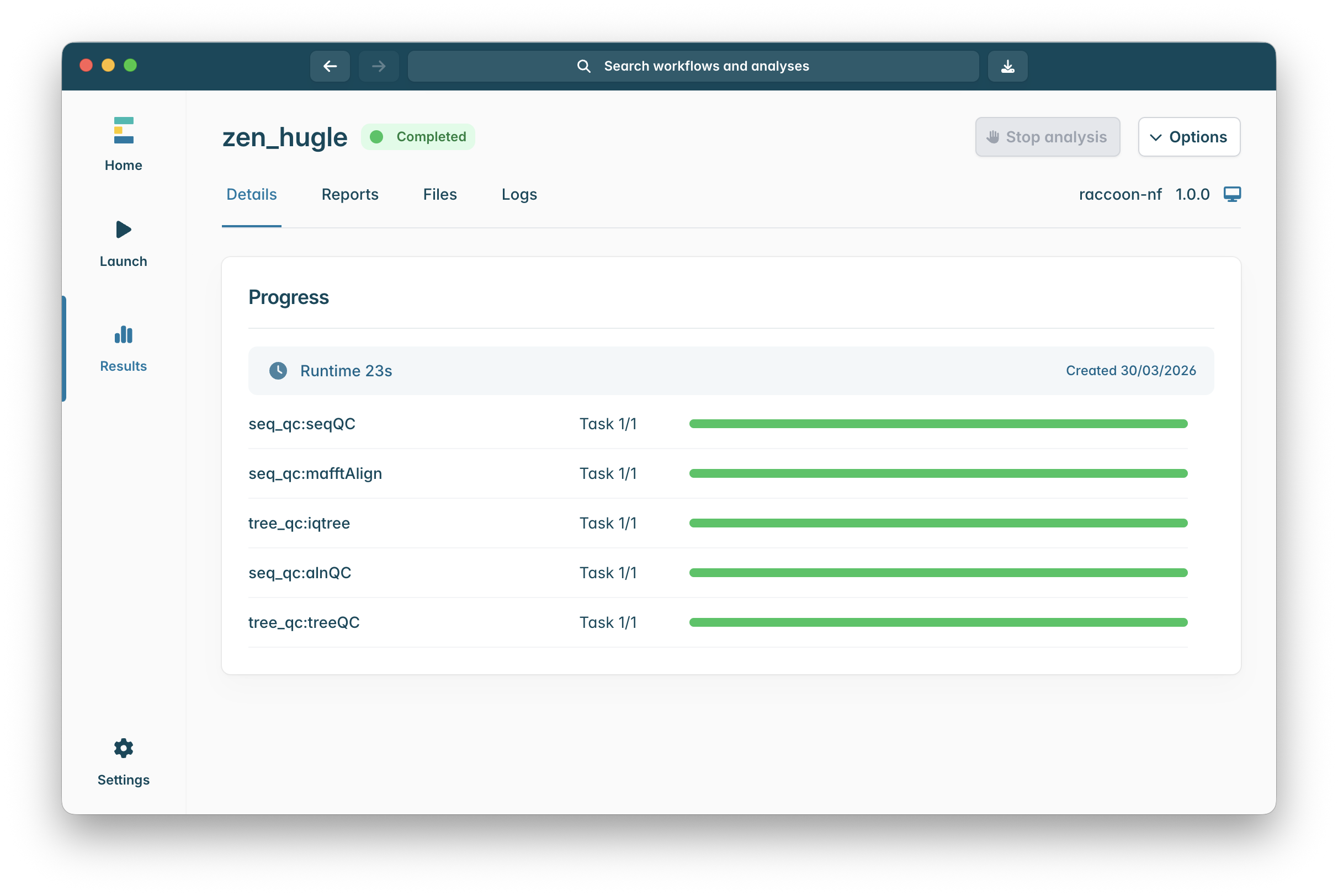
5. Examining raccoon-nf output reports and files
Once the workflow has finished, click on Reports to show the output report documents. The first three reports are from EPI2ME about how the workflow ran - unless there is a problem, these can be ignored.
The last three reports are the outputs of the various stages of raccoon-nf focusing on quality control (qc):
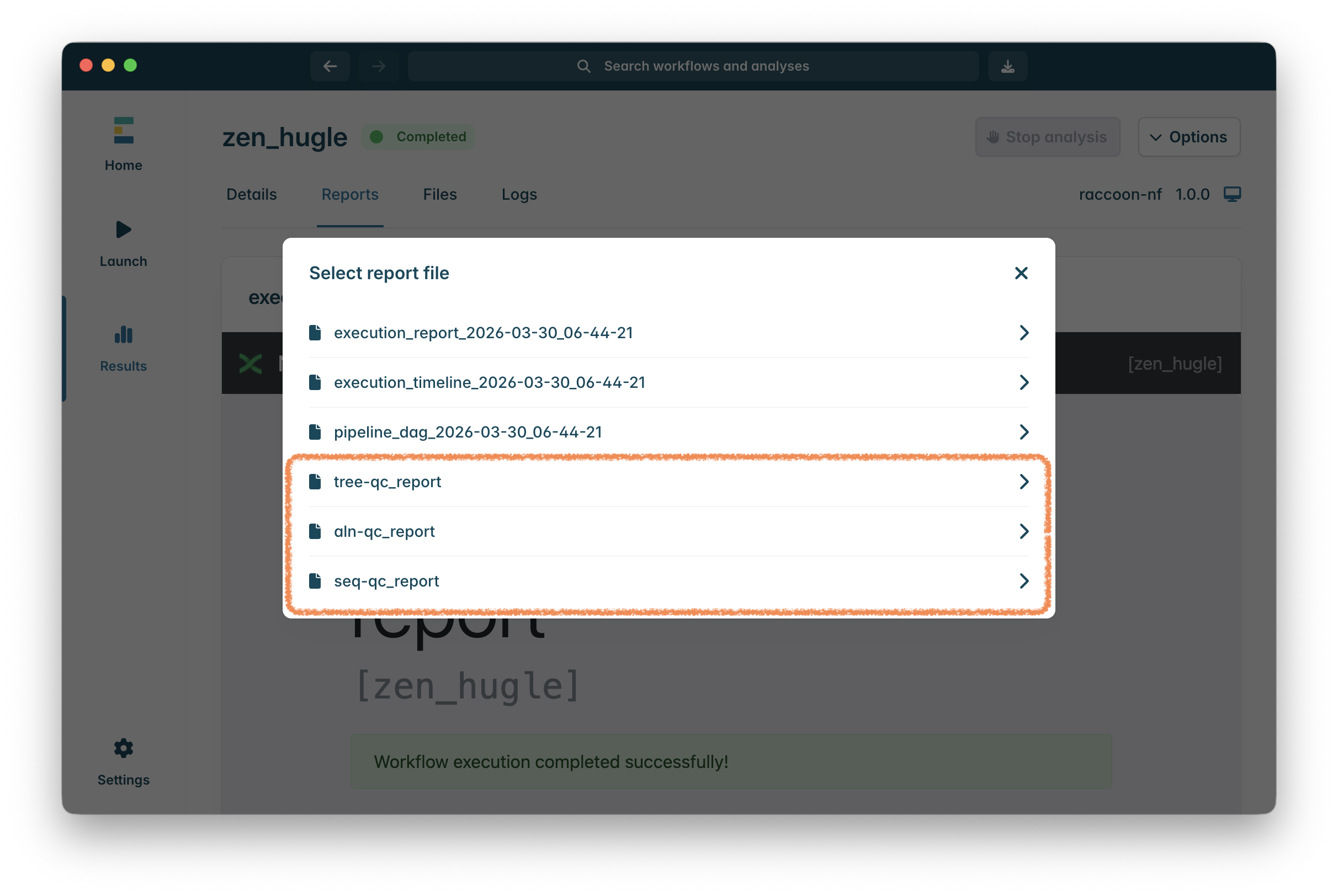
-
seq-qc_report: QC report on the input sequences to find possible sequencing or assembly issues. -
aln-qc_report: QC report on the multiple sequence alignment which identifies sequences with insufficient data to do phylogenetic analysis (these will have been removed automatically). -
tree-qc_report: The final phylogenetic tree report.
If you select the tree-qc_report option you will be able to view the report in the EPI2ME window:
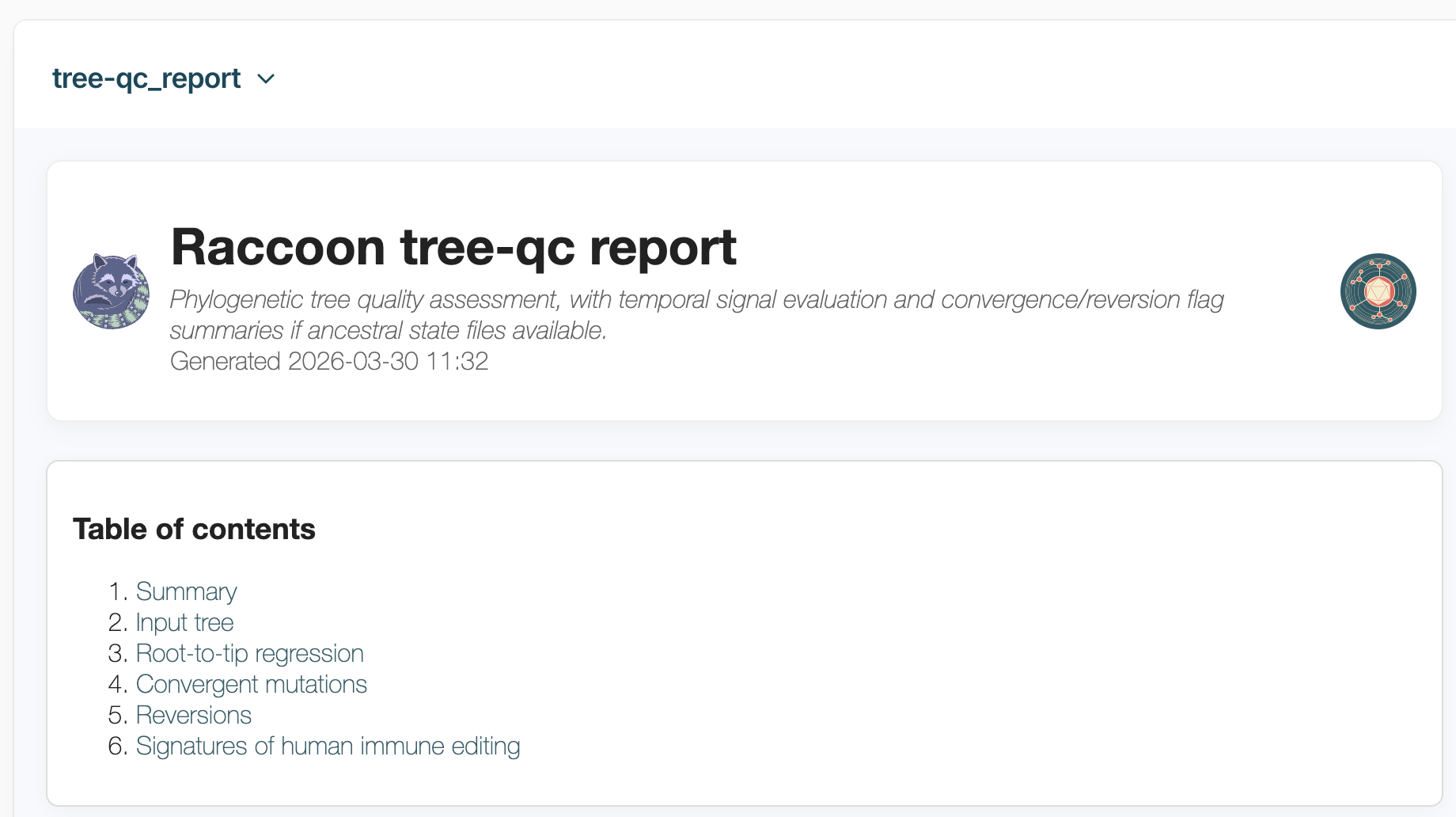
Browse these file and look at the various sections.
For more information about these reports see the full raccoon-nf tutorial:
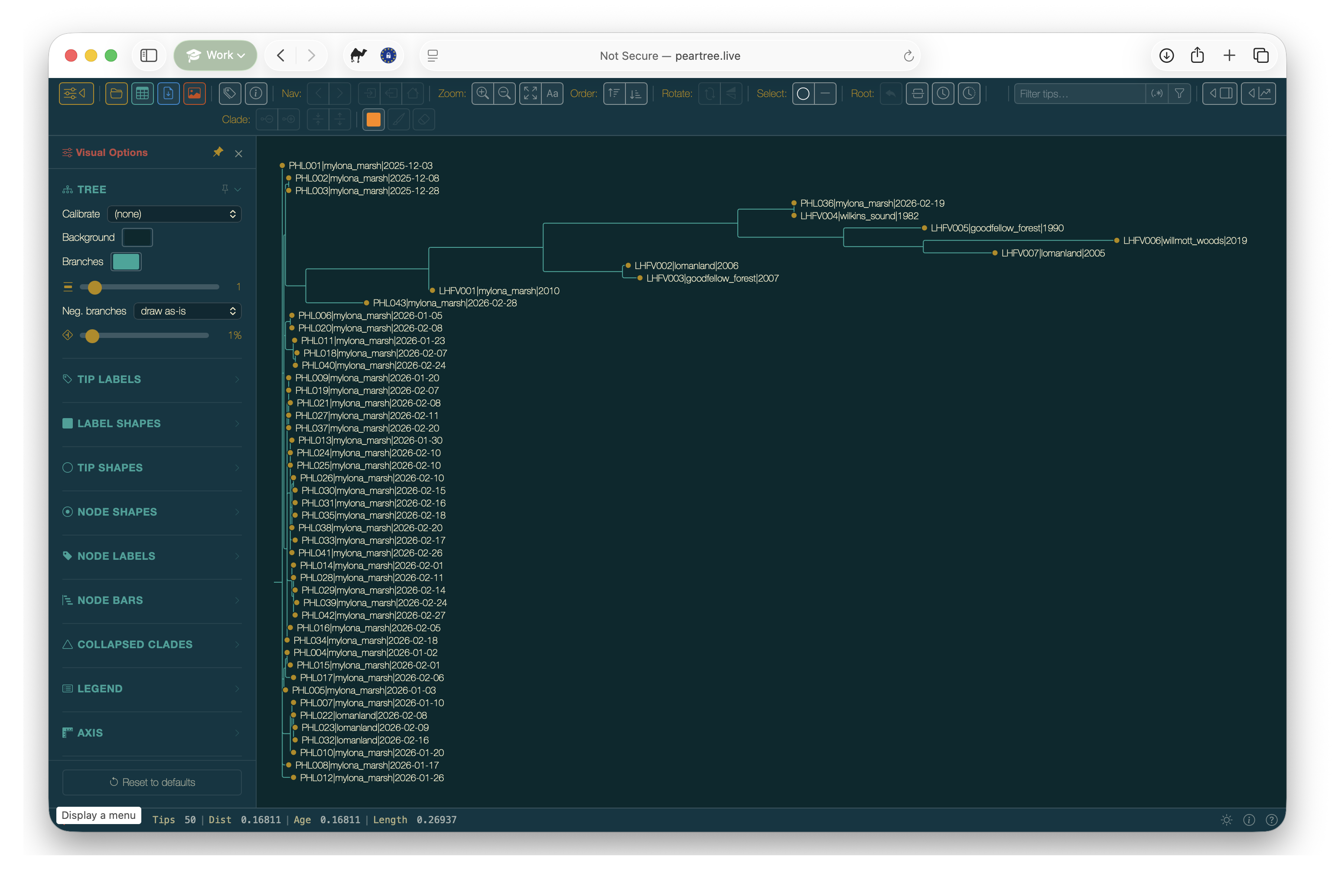
6. Opening the phylogenetic tree in PearTree
raccoon-nf produced a phylogenetic tree file that we can explore in more detail using a tool called PearTree. This is available as a webtool at: http://peartree.live or you can download desktop apps for most operating systems from here: PearTree Downloads.
Find the tree file that was generated by raccoon-nf by clicking on the ‘Files’ tab in your EPI2ME window. Click on all_samples then tree and click the ... on the line labelled all_samples.aln.fasta.treefile:
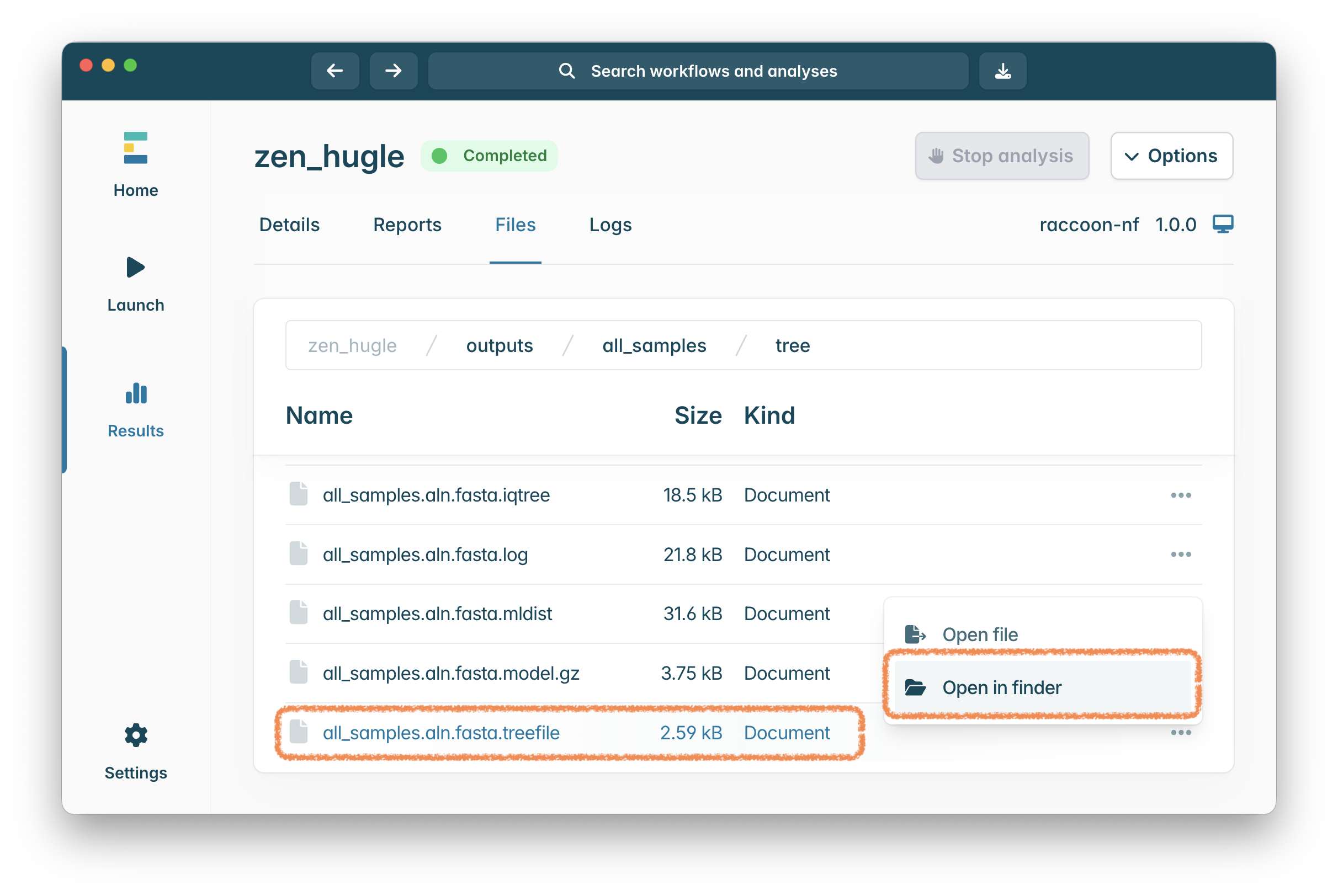
Finally drag the file all_samples.aln.fasta.treefile into the PearTree webpage:
The tree will now open in PearTree ready for investigation: