Multiple sequence alignment and phylogenetics pipeline using raccoon-nf
Raccoon | phylogenetics
| Document: | ARTIC-raccoon-nf-tutorial-v1.0 |
| Creation Date: | 2026-03-15 |
| Last Updated: | 2026-03-15 |
| Author: | Áine O'Toole, Kate Duggan & Daniel Maloney |
| Licence: | Creative Commons Attribution 4.0 International License |
Table of Contents
- Prerequisites
- Background
- Learning outcomes
- Setup
- 1. Understanding and exploring the datafiles
- 2: Understanding raccoon-nf pipeline
- 3. Running raccoon-nf in EPI2ME
- 4. Interpreting the output of raccoon-nf
- Discussion
- Additional Resources
Prerequisites
This tutorial assumes the Epi2Me software is installed and running and the raccoon-nf workflow has been installed:
Background
Working with multiple FASTA files and creating clear, informative headers for phylogenetic analysis can be much easier with the right tools and guidance. For those who may be less familiar with coding or the command line, tasks such as merging FASTA files or structuring metadata-rich headers can be time-consuming, especially when done manually. This tutorial introduces practical approaches that simplify these steps and help reduce the chance of errors in metadata or sequence entries.
In addition, understanding and critical evaluation of the quality of results is an important part of viral phylogenetics. This tutorial will guide you through sequence quality-control steps, highlight potential issues that can arise during multiple sequence alignment, and help you confidently assess and interpret the resulting phylogenetic trees. An interactive description of the pipeline can be found at artic-network.github.io/raccoon/docs/interactive_pipeline
In this tutorial, the pipeline will be run using the EPI2ME user interface. Users do not need to run every command manually, and do not need knowledge of the command line to run raccoon, however they should understand each stage. Users that are familiar with the command line can run different modules independently in their own custom workflows to aid in post-consensus analysis QC.
Learning outcomes
By the end of the session, participants should be able to:
- Explain why metadata harmonisation is essential before phylogenetic analysis.
- Identify common metadata and sequence quality problems and describe their downstream impact.
- Understand the importance of alignment curation, and the impact of alignment issues on phylogenetic inference.
- Appreciate the importance of background data and sampling bias.
- Interpret key outputs from raccoon modules
seq-qc,aln-qc, andtree-qc. - Critically assess root-to-tip plots, read a phylogenetic tree structure, and understand phylogenetic structure that may indicate upstream analytical issues.
1. Understanding and exploring the datafiles
Concepts to cover
- FASTA and metadata formats
- The content of the provided files
Steps
A FASTA-formatted file contains sequence records, which can be amino acid or nucleotide sequences. A record minimally contains two pieces of information:
- The sequence ID (e.g. PP00001)
- The sequence itself (e.g. CGATCGAT…ACTGACT)
Format details:
- The sequence ID is stored in the header line, denoted by a
>symbol - The header line may also contain additional information (sequence description) after the first space
- Important: The sequence ID must not contain whitespace (spaces or tabs)
- The sequence is stored on the following line(s)
- Sequences can be split across multiple lines for readability
- The next record does not start until the next line that begins with
>
a)
>PP00010 barcode=barcode01 AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACGAGCTAGCTAGCGTAGCTAGCGCATTACGTACTACGAGCTAGCTAGCGTAGCTAGCGCATTACGTACTACG b)
>PP 00011AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACGGGCTAGCTAGCGTAGCTAGCGCATTACGTACTACTTGCTAGCTAGCGTAGCTAGCGCATTACGTACTACAACGTAGTCATAGTCGTACTGACc)
PP00012AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACGd)
>PP00013|inis_aine|2026-03-16AGCTAGCTAGCGTAGCTAGCGCATTACGTACTACGAGCTAGCTAGCGTAGCTAGCGCATTACGTACTACG
In order to properly inform our analysis, we need to integrate our sequence data with sequence metadata. Metadata is data that provides additional information about our samples, such as collection date or location. This is usually supplied as an additional file in CSV or TSV format.
Depending on the data collection process, planning, and ethics approvals, metadata may be very detailed or more sparse.
Location, immune status,travel history, sample collection date, ct value, symptoms, symptom onset date, gender, age, occupation, patient eye colour, patient height
We are also providing metadata files to accompany the sequence data. Much of the work in preparing metadata files has already been done, however use these files as a guide for future analyses.
Download and unzip the provided files: raccoon_tutorial_data.zip
If you have carried out a sequencing run as part of the workshop and have new case data, use that file. Otherwise, a FASTA file of case data can be downloaded from here.
2: Understanding raccoon-nf pipeline
Concepts to cover
- What steps are run as part of the raccoon-nf pipeline
Pipeline details
Running best-practice phylogenetics can be challenging, however with the raccoon-nf pipeline a simple alignment and phylogenetic workflow can be performed in a single step. The pipeline itself is configurable, however in this tutorial we will be running the steps shown in the figure below.

A) Input files
- input sequences (one or more fasta files or directory containing fasta file)
- input metadata (one or more metadata files (csv or tsv) or directory containing metadata files)
B) raccoon seq-qc
Outputs:
- a combined fasta file with sequence headers harmonised and populated from the metadata fields
- seq-qc_report.html (a report describing the dataset, the matching, the output and any issues identified with the data)
- seq-qc_filter_failures.csv (sequences that do not pass qc filters, max n and min length)
- seq-qc_metadata_issues.csv (flagging missing metadata fields or sequences that failed to match metadata)
C) alignment
Multiple sequence alignment is a key step prior to running phylogenetics. It is the scaffold upon which we can begin to reconstruct the evolutionary relationships between different sequences in the tree. We will run alignment using MAFFT, which is a popular software tool for creating multiple sequence alignments.
Output:
- An aligned fasta file
D) raccoon aln-qc
A high-quality alignment is crucial to generating a good phylogenetic tree. Being able to accurately assess whether there are issues with your multiple sequence alignment is a key skill that we will cover today.
The alignment is checked for various issues that may impact the quality of the phylogenetic inference. Different kinds of SNPs (clustered SNPs, N-adjacent SNPs, gap-adjacent SNPs) are flagged that may suggest issues with the alignment or with a given sequence. If a given sequence has many issues flagged (default >20), that sequence is flagged for removal from the analysis. Flagged SNPs do not necessarily mean there is anything wrong with the SNP, it may reflect genuine biological variation. However, these sites may need to be investigated closely.
Output:
- aln-qc_report.html (a report describing the input alignment, n content and any SNPs that were flagged as possibly pro)
- mask_sites.csv (describes the sites flagged for investigation or masking and the sequences flagged for removal)
E) tree estimation Tree building is run using IQTREE. The substitution model used is configurable and an outgroup can optionally be included. If an outgroup is included, ancestral state reconstruction will be run during the tree building process to provide additional checks on the tree, and the outgroup sequence will be pruned off from the final tree. In this case, as we are not yet familiar with the data, we will not select an outgroup as it is not clear what an appropriate outgroup would be.
Key output:
- *.treefile (a maximum likelihood tree file)
F) raccoon tree-qc
Output:
- tree-qc_report.html (report showing the tree, a root to tip and any issues that were flagged during the tree-qc process)
- *.phylo_flags.csv
- A midpoint rooted tree (if no outgroup provided)
- Branch reconstruction file (if outgroup provided)
- State difference file (if outgroup provided)
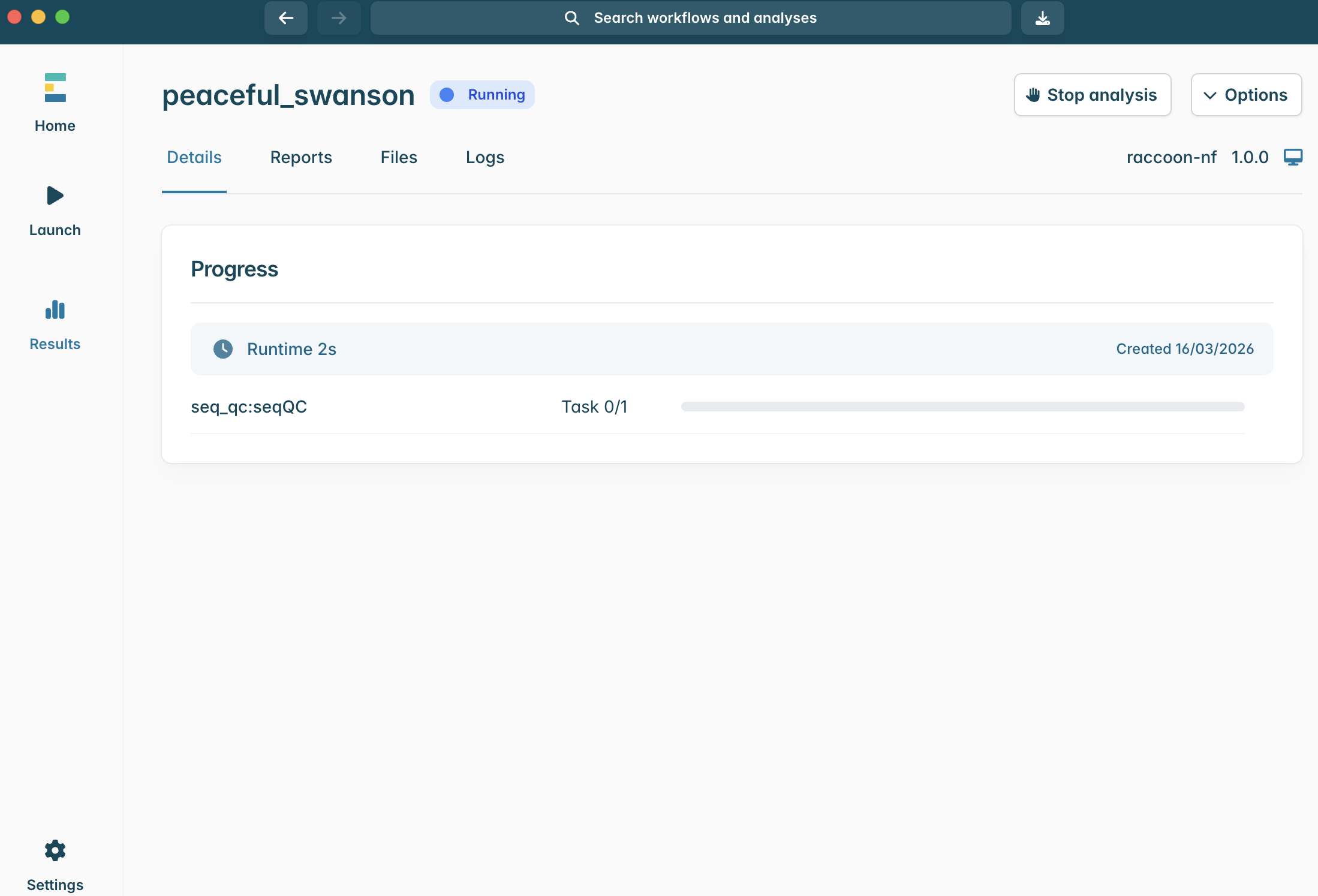
3. Running raccoon-nf in EPI2ME
Concepts to cover
- Launching a workflow in EPI2ME
-
Configuring the raccoon-nf workflow settings to match the input data
Steps
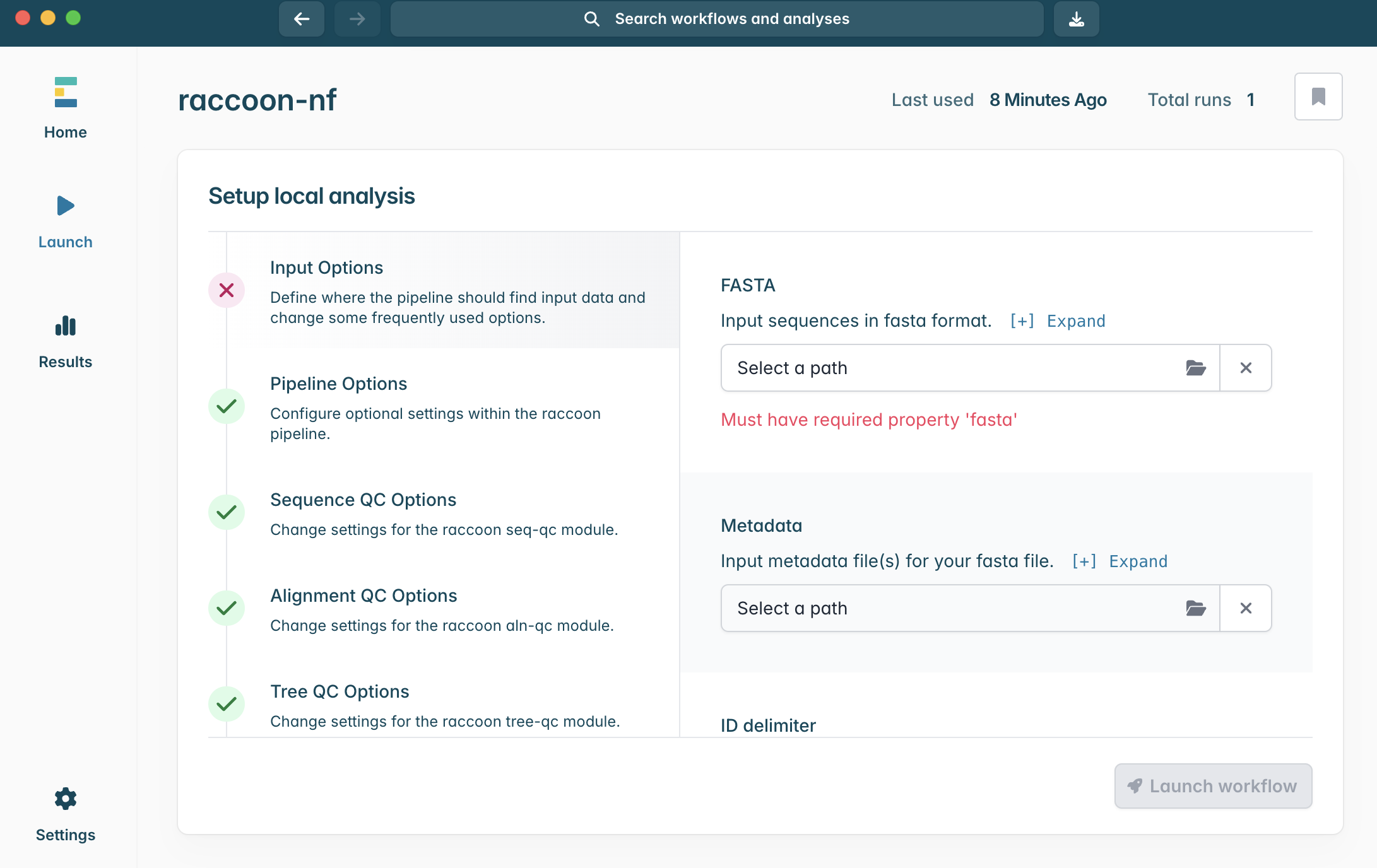
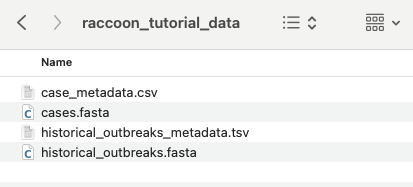
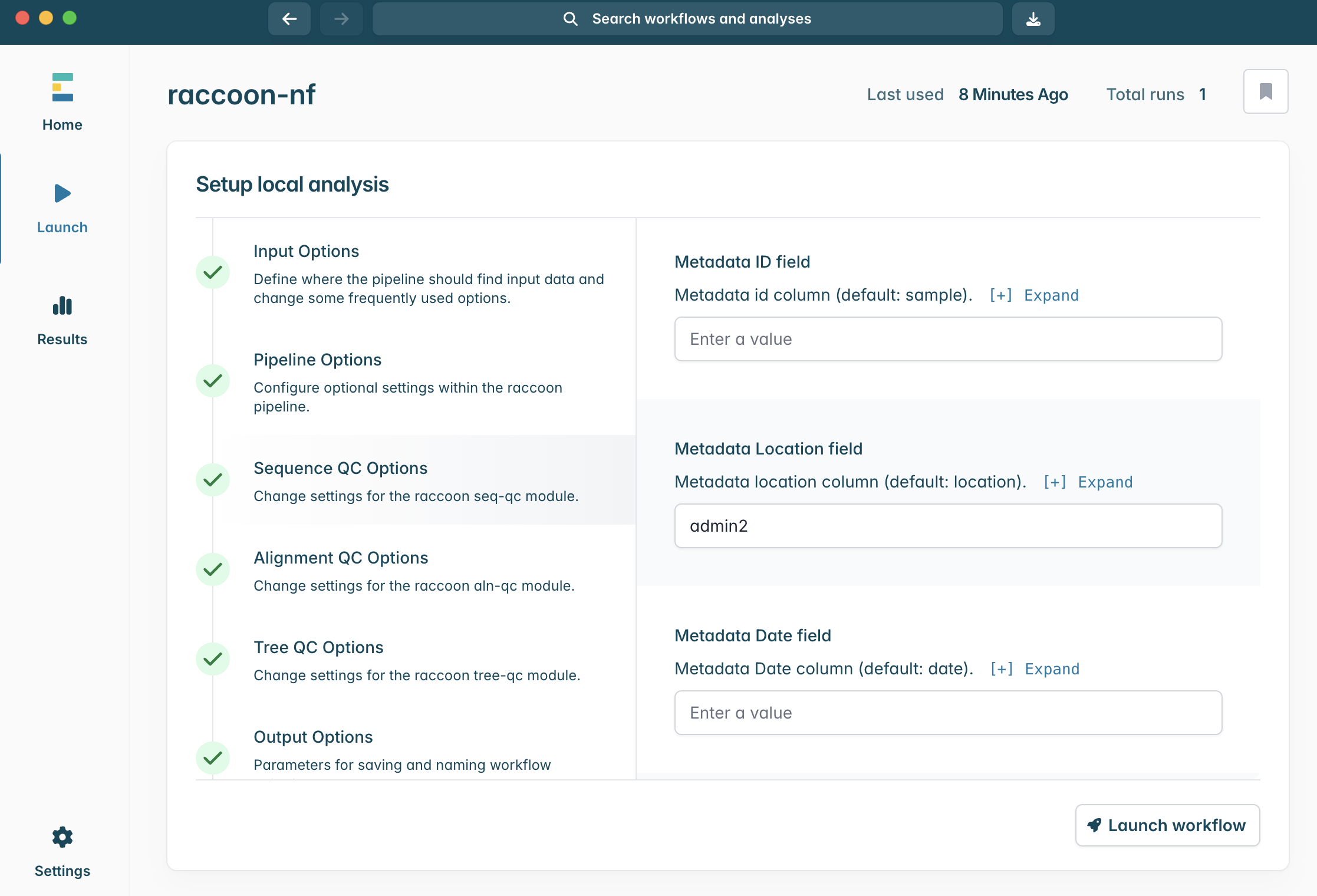
Input Options panel, select the directory that contains your FASTA files (it should have unzipped into a directory called “raccoon_tutorial_data”), and select the same directory for your metadata files. Raccoon-nf will automatically detect which files present are FASTA files and which ones are metadata files based on the file extensions (i.e. .fasta or .csv/.tsv). We can leave the remaining Input options as default.
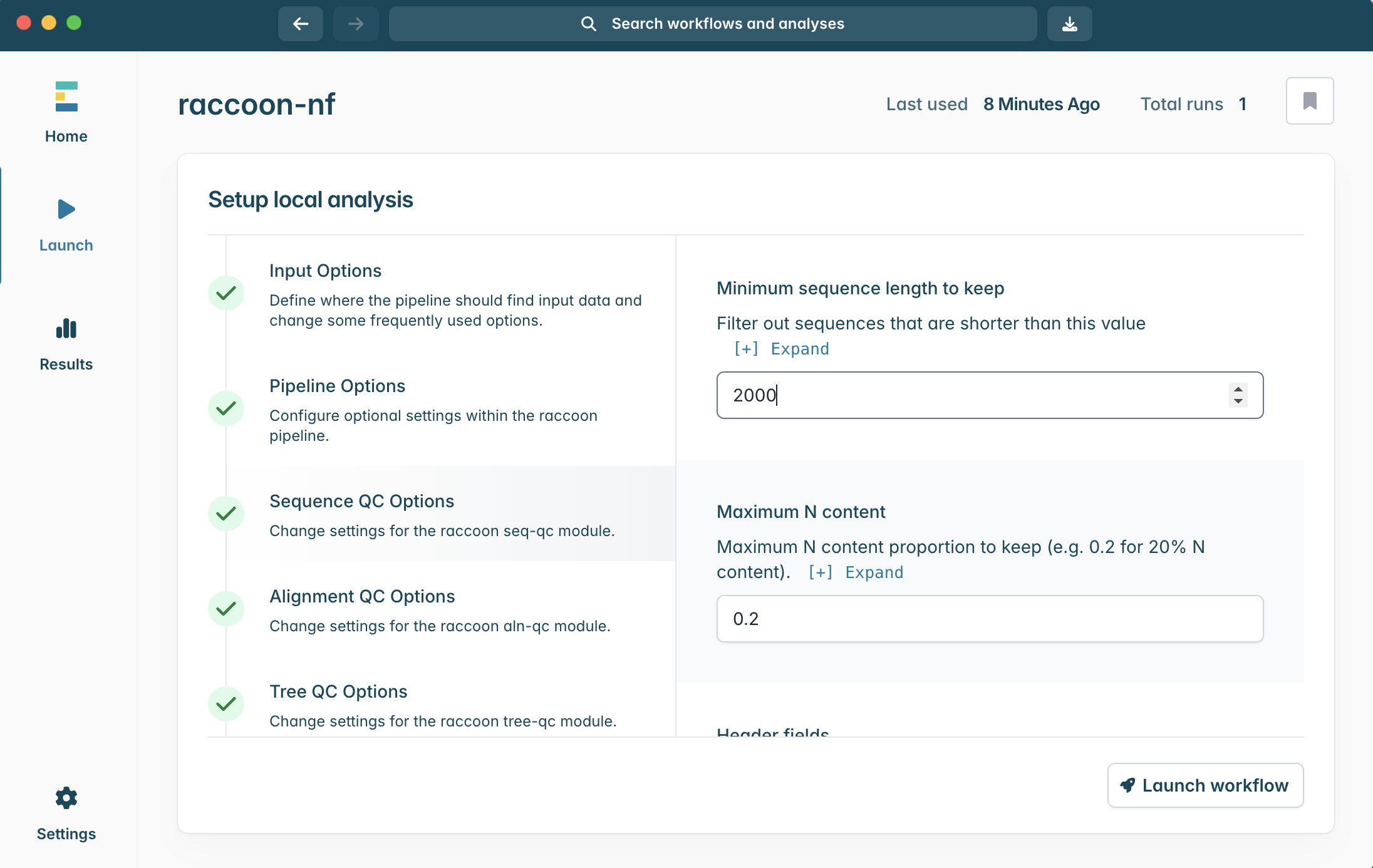
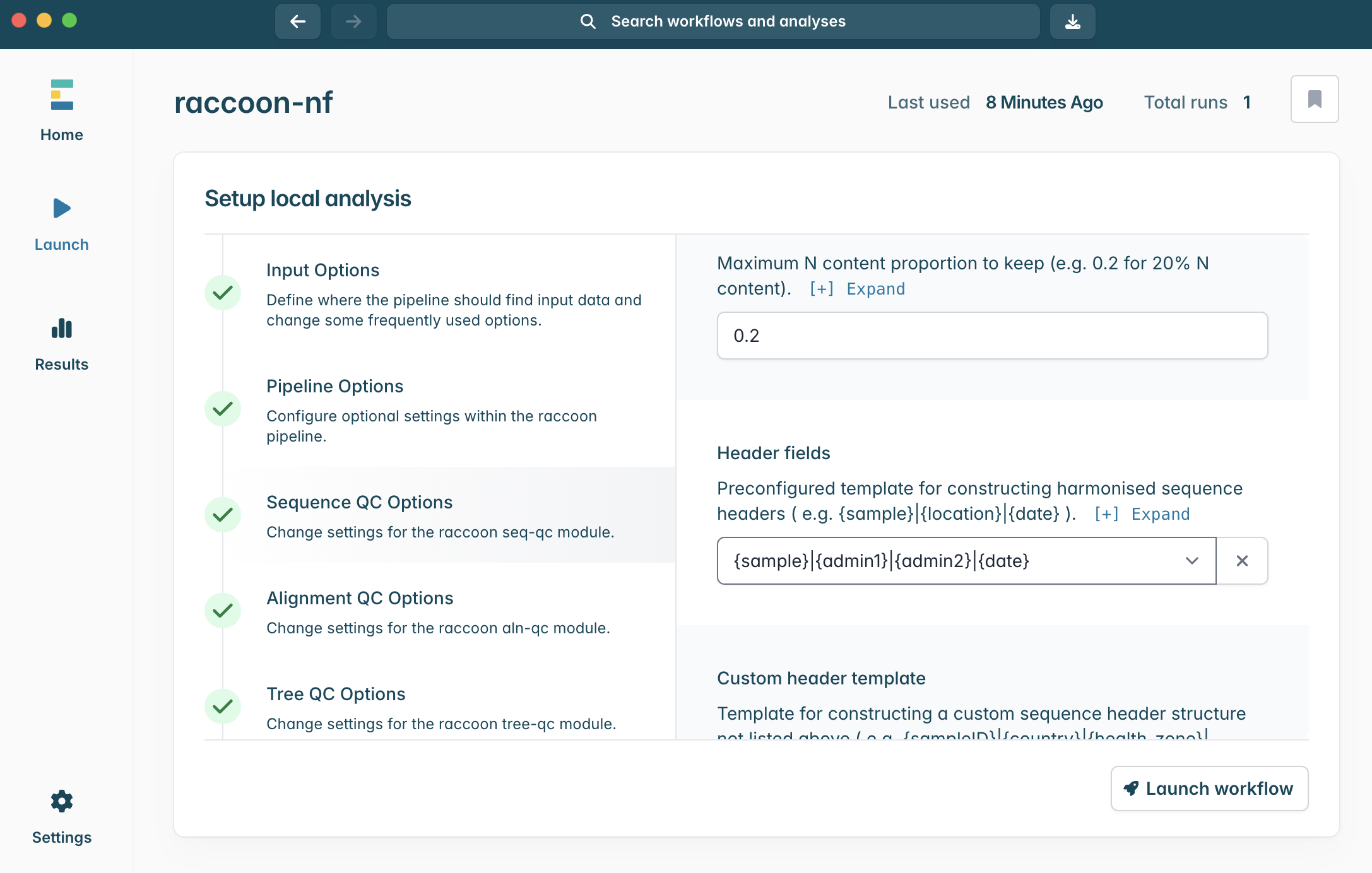
1.
{sample}|{date}2.
{sample}|{admin1}|{admin2}|{date}3.
{admin2}|{date}4.
{sample}|{location}|{date}
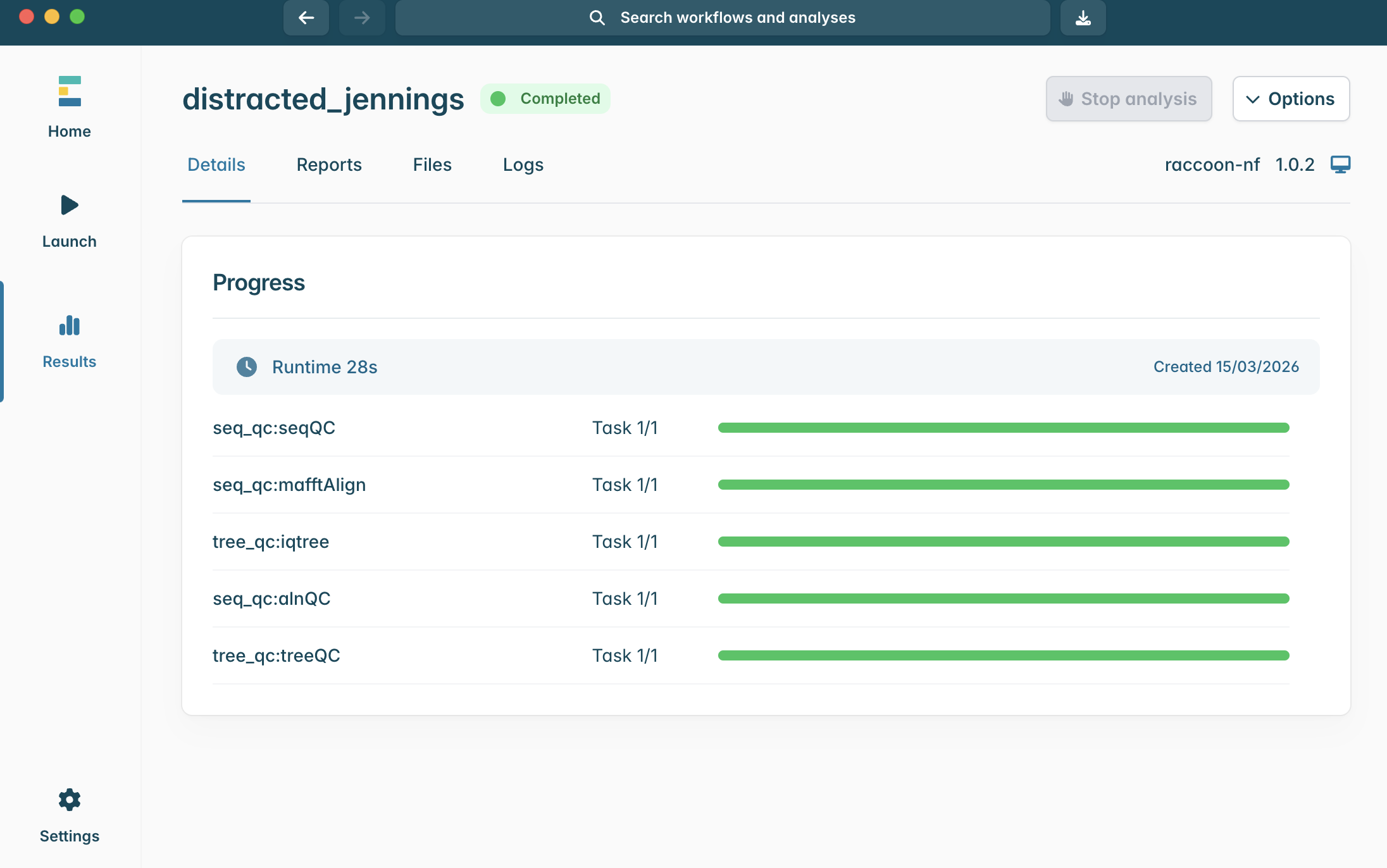
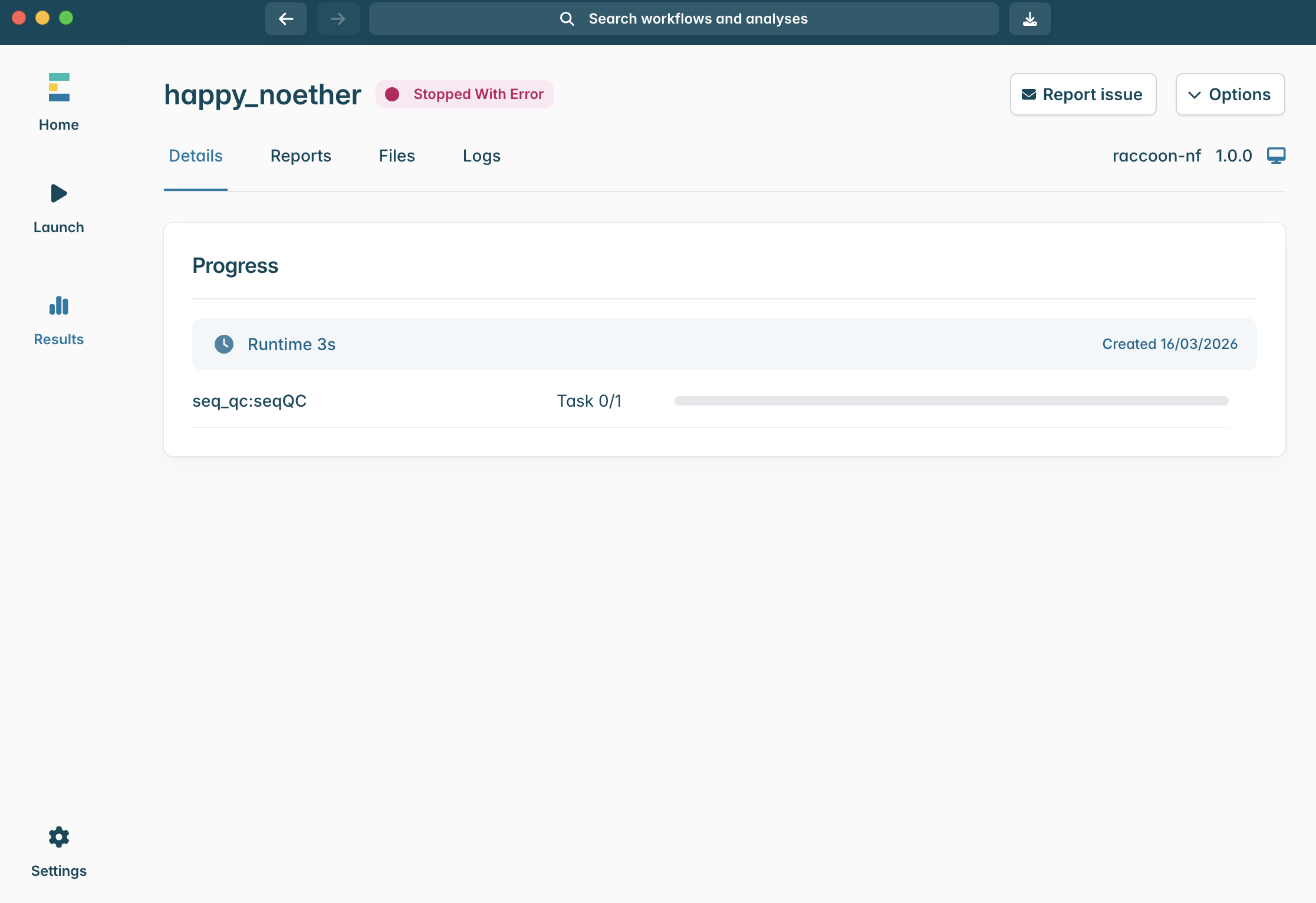
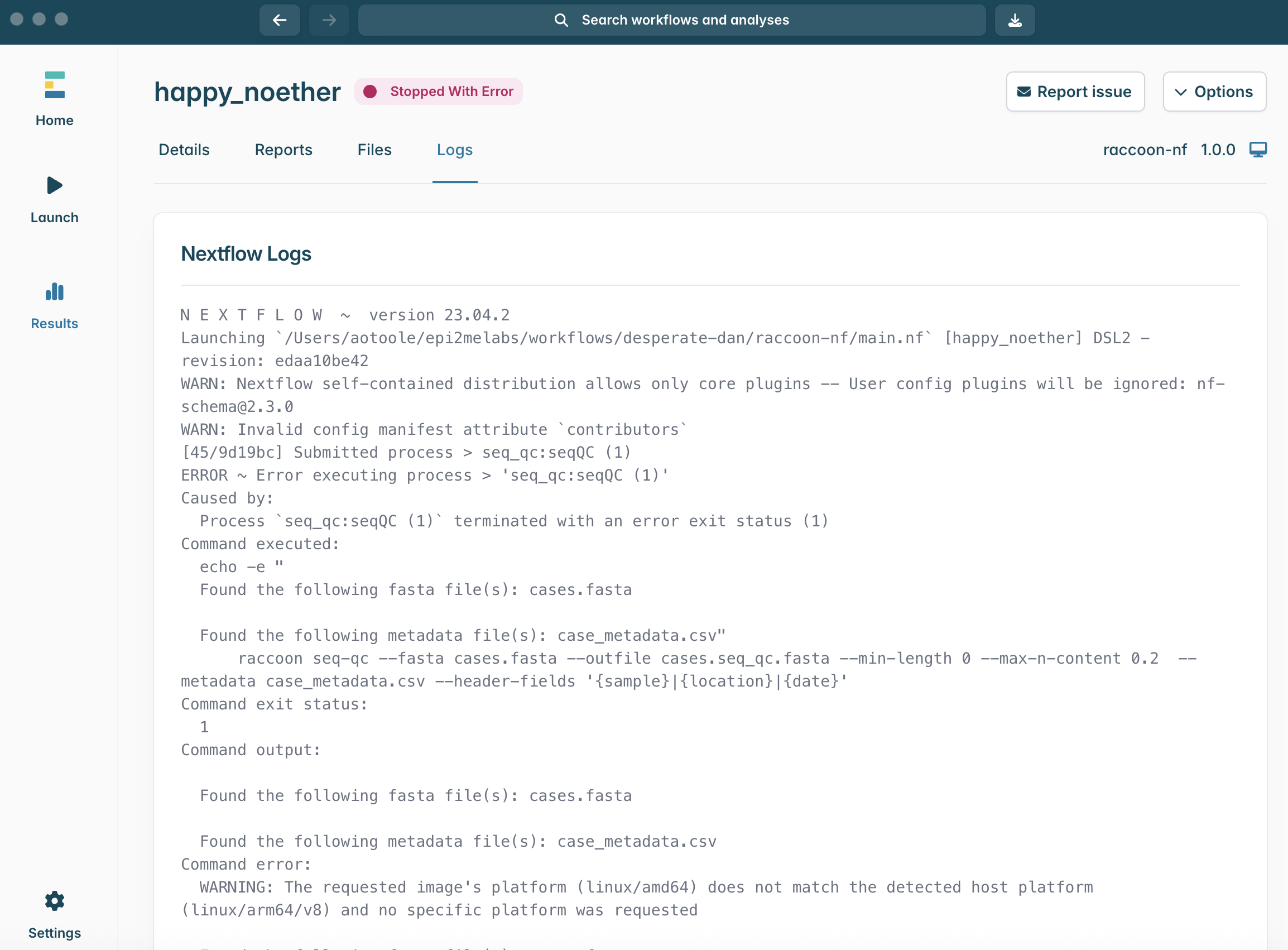
ERROR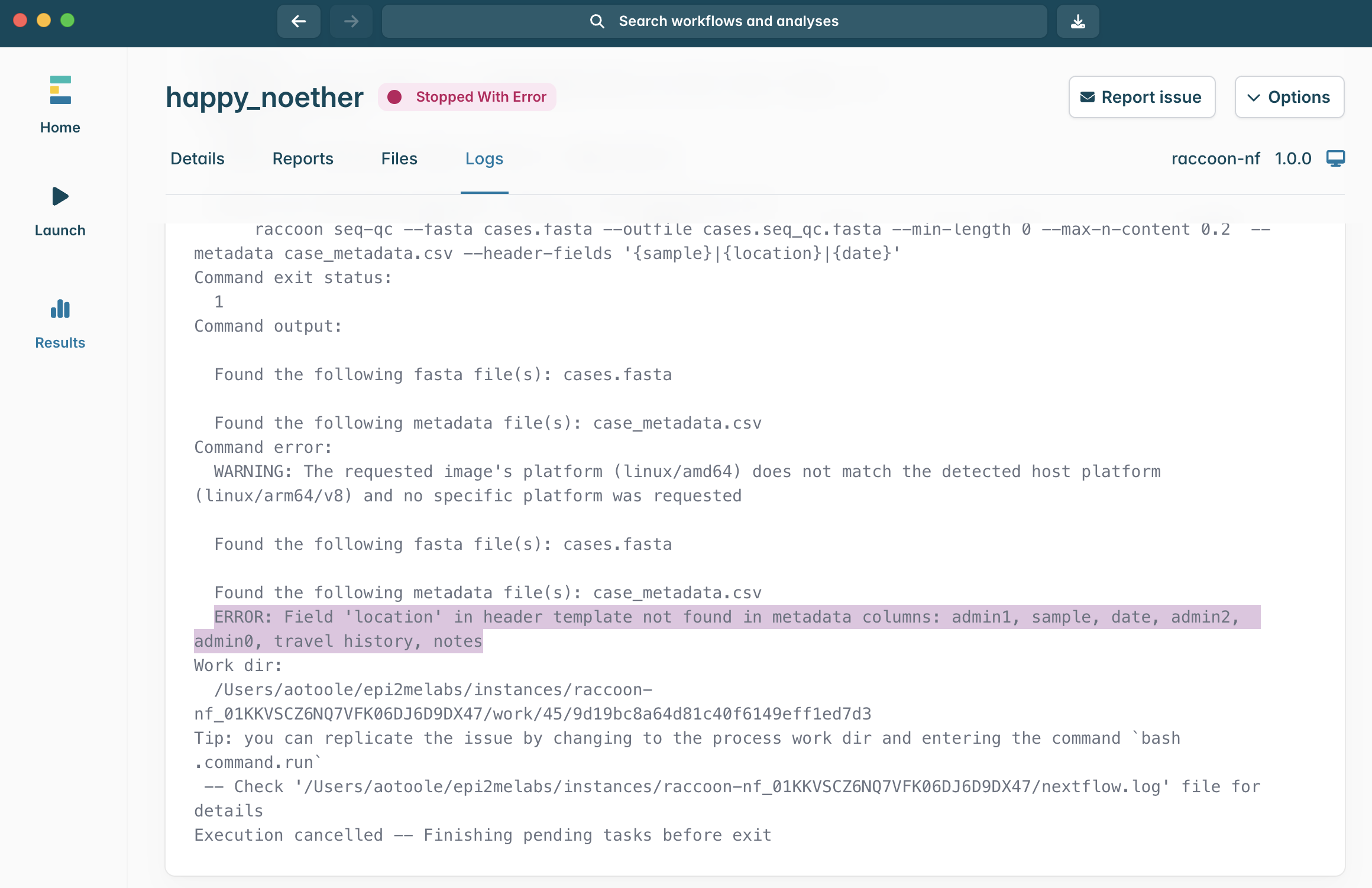
Checkpoint questions:
- What criteria are used to filter sequences?
- If our metadata file contained the following columns – ID, country, health_zone, sample_date – what would an appropriate header fields template be?
- Where can you find information that can help explain an error?
4. Interpreting the output of raccoon-nf (seq-qc)
Concepts to cover
- ID matching between sequence headers and metadata table rows
- Harmonising metadata from multiple files
- Preserving epidemiologically useful fields in headers
Steps
The report summary section provides an overview of the data including total sequences, the date range associated with the sequence metadata and any filters applied.
The inputs files section describes the FASTA and metadata files loaded into raccoon, with an overview of the number, length and ambiguous base content of the sequences in the input FASTA files. Click to expand each file to view QC information for each sequence.
The sequence length distribution plot shows lengths of the sequences in the input FASTA files.
The metadata description plot summarises the geographical and temporal distribution of the dataset provided.
The filtered sequences table highlights sequences flagged for filtering during the QC check.
The final dataset table describes the final, combined dataset, with harmonised headers.
Finally, any metadata issues such as missing fields or inability to match sequences are flagged in the metadata issues table.
Discussion
- Is it better to relax
--max-n-contentor drop marginal sequences? Why? - What are the risks of including poor-quality but epidemiologically important samples?
4. Interpreting the output of raccoon-nf (aln-qc)
Concepts to cover
- The impact of high N content on downstream phylogenetic inference
- The importance of visually inspecting your alignment
Steps
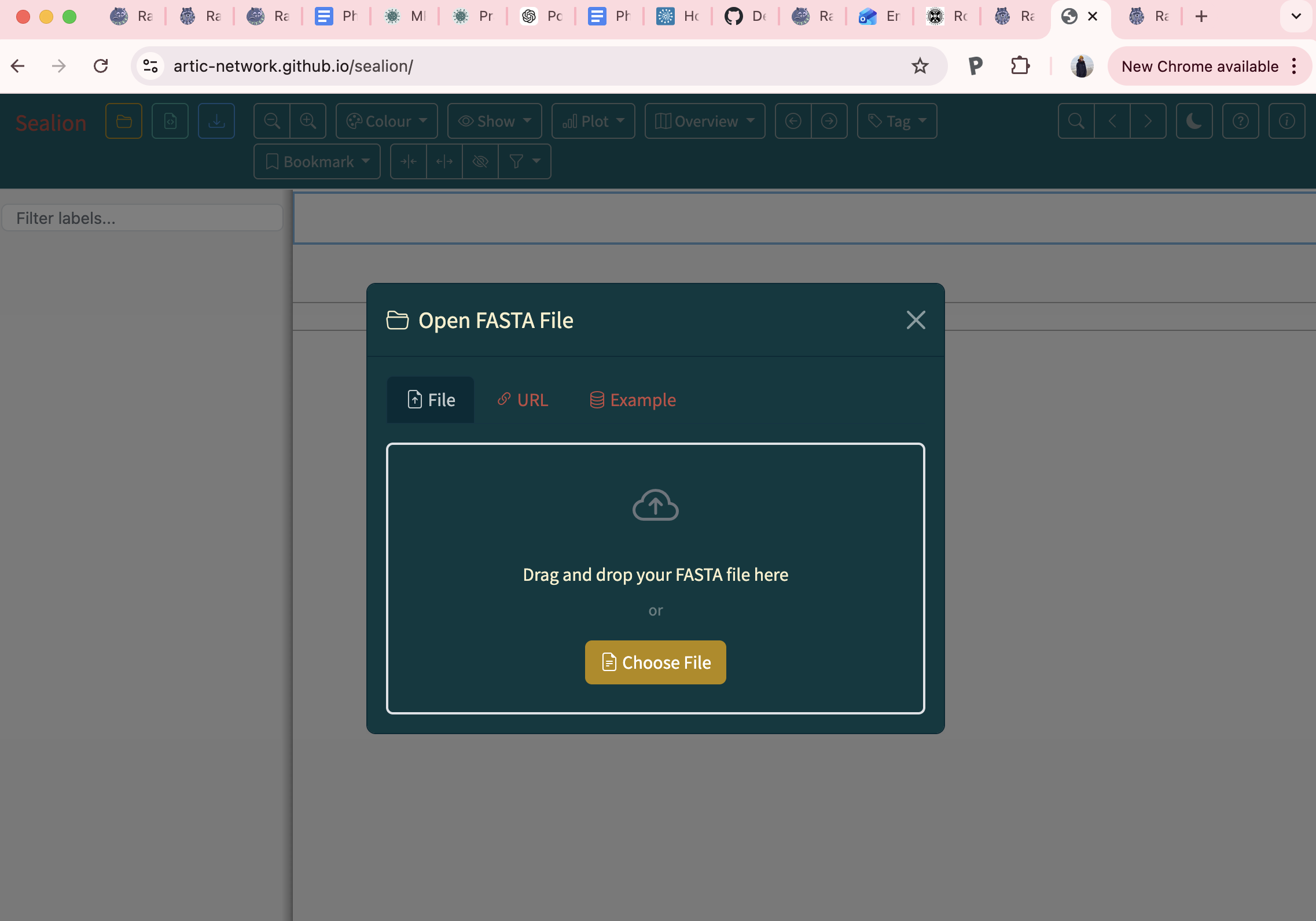
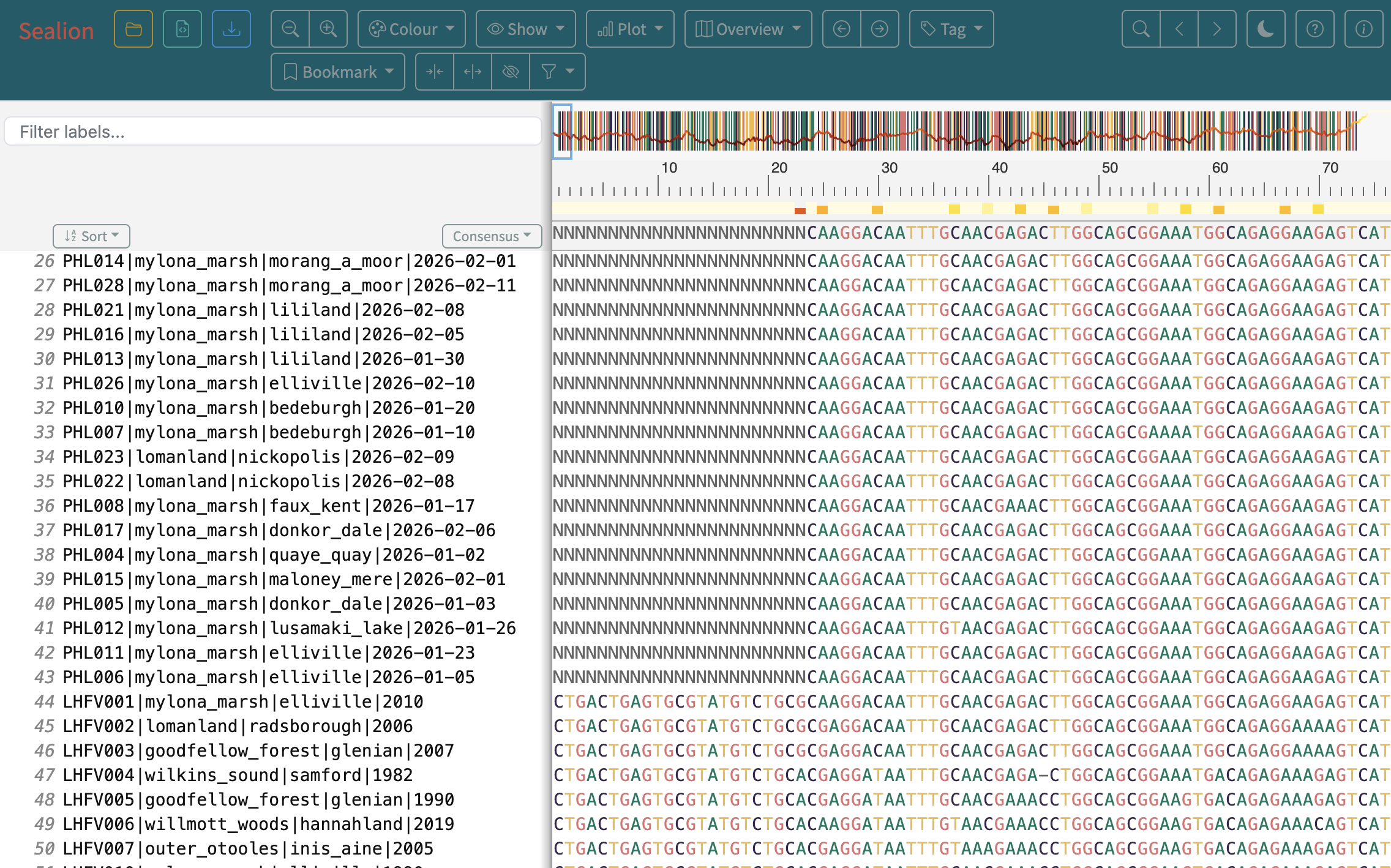
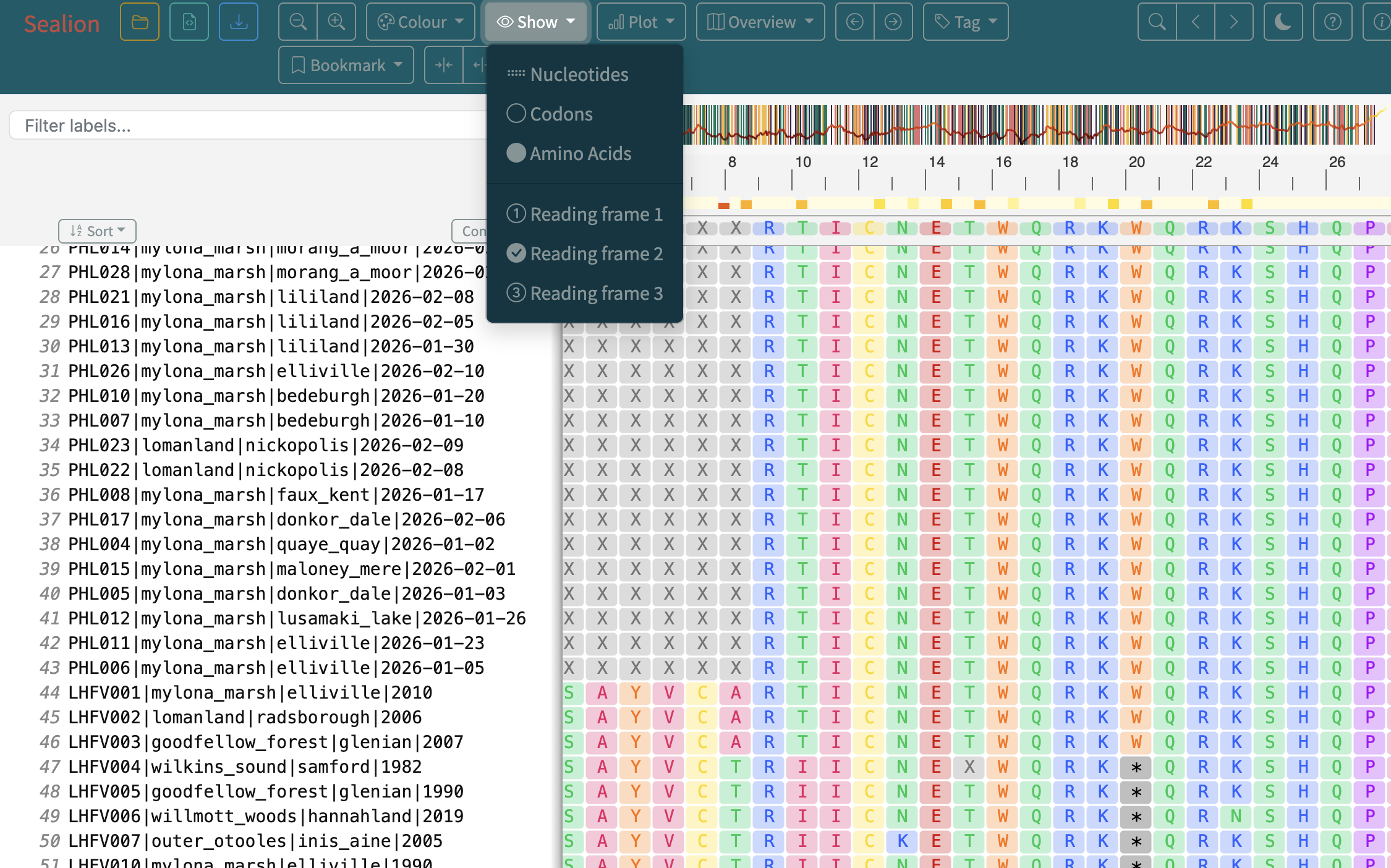
4. Interpreting the output of raccoon-nf (tree-qc)
Concepts to cover
- How to read a phylogenetic tree
- Interpreting a root-to-tip plot
Steps
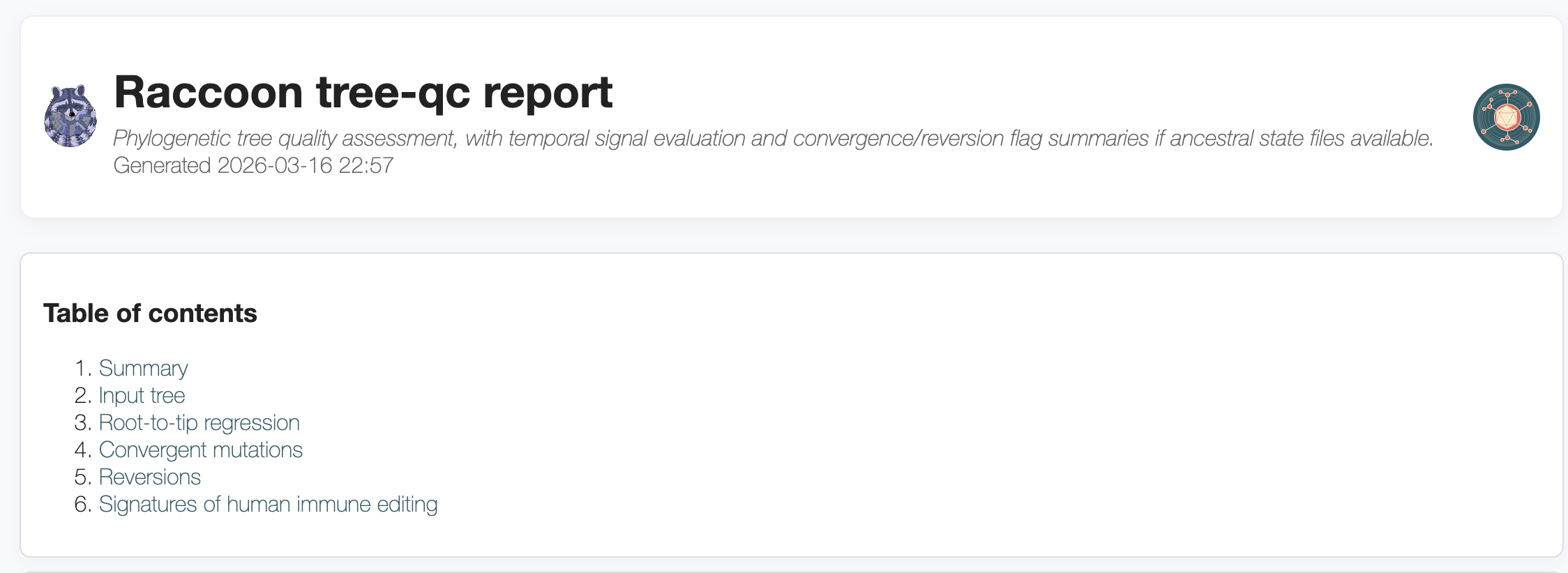
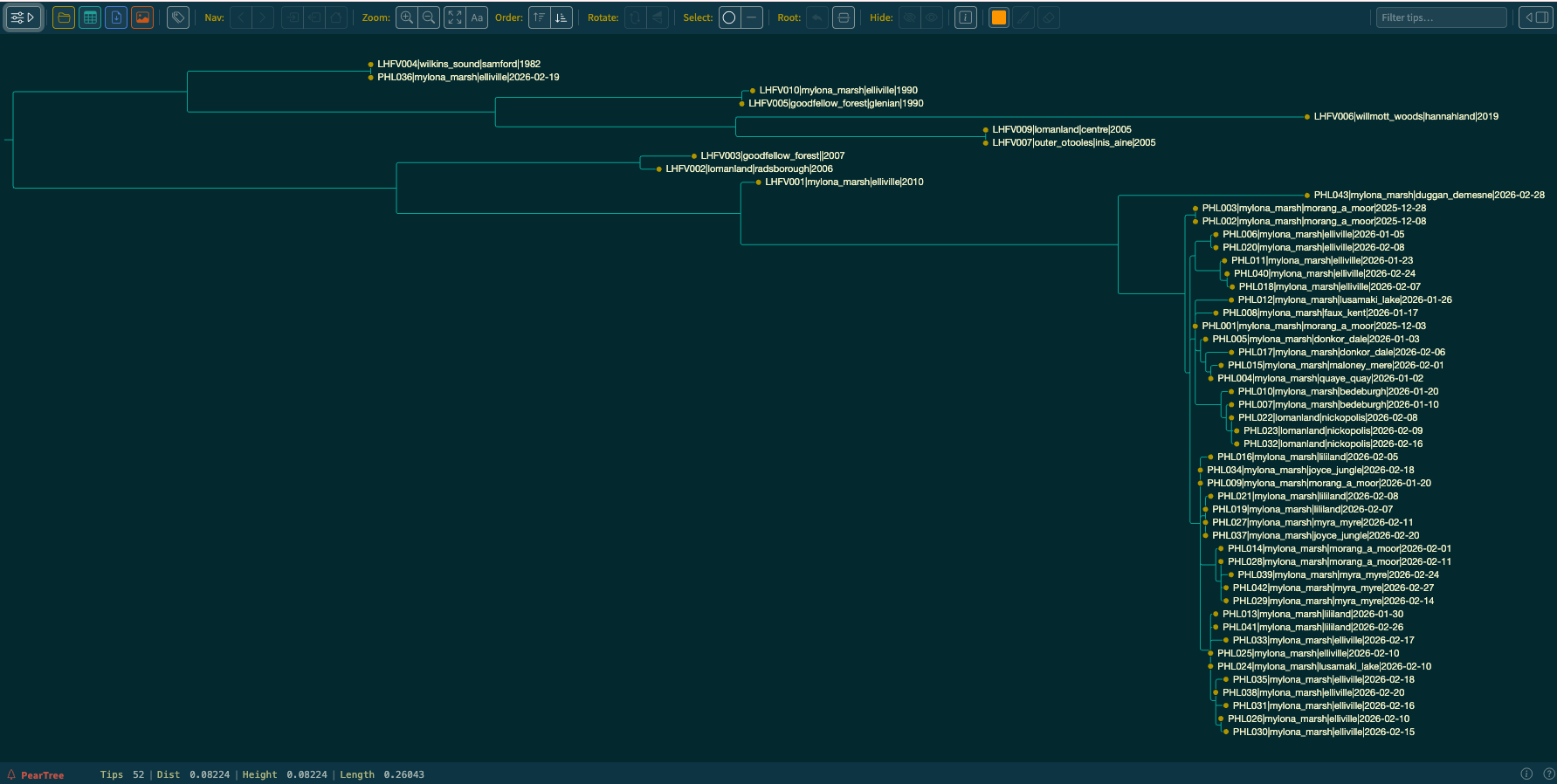
tree and identify the .treefile file.Discussion
Additional Resources
Software used in raccoon-nf
Attribution
This tutorial is intended for both:
- workshop delivery (guided teaching)
- self-paced reference for raccoon users
