Running the MPXV alignment and phylogenetics pipeline using Epi2Me
Squirrel | phylogenetics
| Document: | ARTIC-MPXV-squirrel-EPI2ME-SOP-v1.0 |
| Creation Date: | 2024-08-21 |
| Author: | Áine O'Tool, Rachel Colquhoun |
| Licence: | Creative Commons Attribution 4.0 International License |
Credits / Acknowledgements
This workflow runs the Squirrel pipeline written by Áine O’Toole. Squirrel itself makes use of a number of open-source software packages, including minimap2, gofasta and IQTREE2, in addition to custom code for APOBEC3 analysis.
Using the Squirrel MPXV phylogenetic analysis pipeline in EPI2ME
Requirements:
- A working installation of EPI2ME. For instructions for installing EPI2ME, see this document.
- Internet access to download the pipeline, and for the first time running it. After that, you should be able to run it offline.
- Details about how the data was generated including the primer scheme used and the base-caller specified within the MinKNOW software.
Rationale
MPXV is a large poxvirus, with a complex dsDNA genome ~200kb in length. Alignment of sequences, and therefore phylogenetics, is challenging using classic due to tracts of low-complexity and repetitive regions. Squirrel provides an efficient map-to-reference alignment pipeline with masking of problematic regions of the genome. Full details of the pipeline and workflow can be found here.
Command line
Squirrel can be used as a command line tool, with full command-line documentation available on the squirrel GitHub repository at (github.com/aineniamh/squirrel)[https://github.com/aineniamh/squirrel].
EPI2ME user interface
Squirrel can also be run through the EPI2ME user interface. Please first install the EPI2ME desktop application using the provided link. You can then go to ‘available workflows’ then ‘Import workflow’ from https://github.com/artic-network/squirrel-nf as shown below:
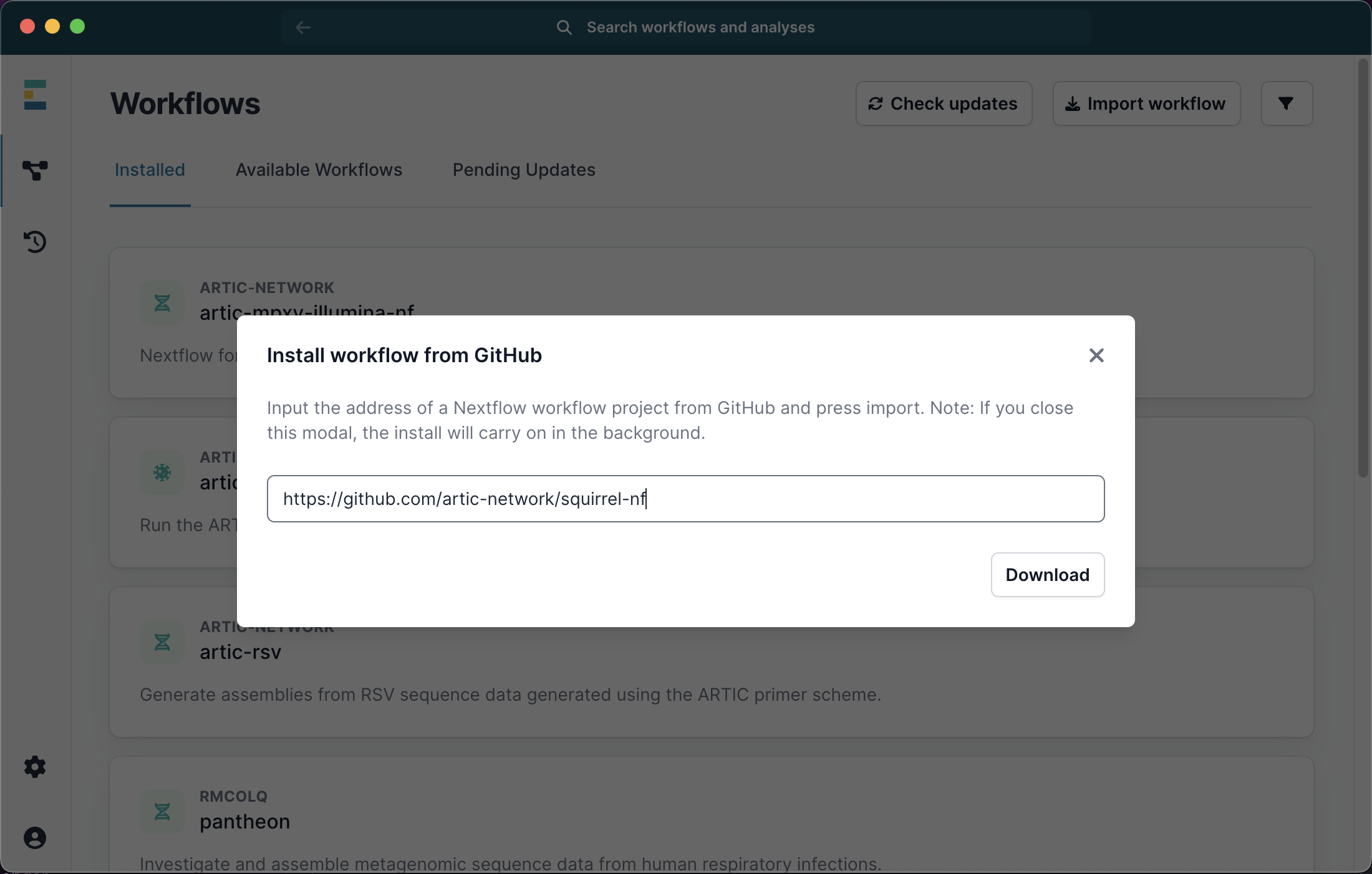
Once the workflow has successfully downloaded, you can click the X to exit to download window, and select it from the list of available workflows. Next select Run this workflow from the available options, and then Run on your computer:
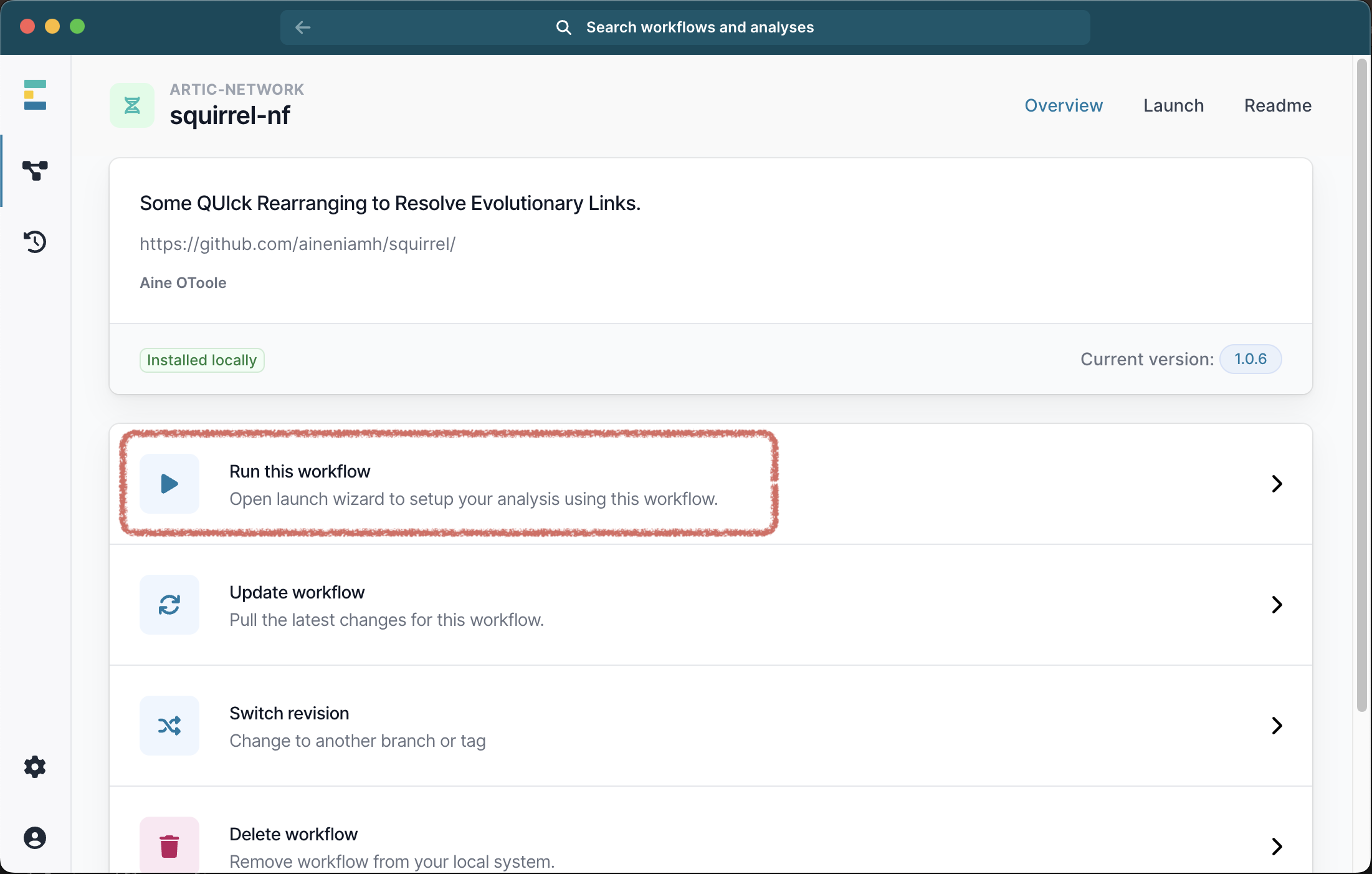
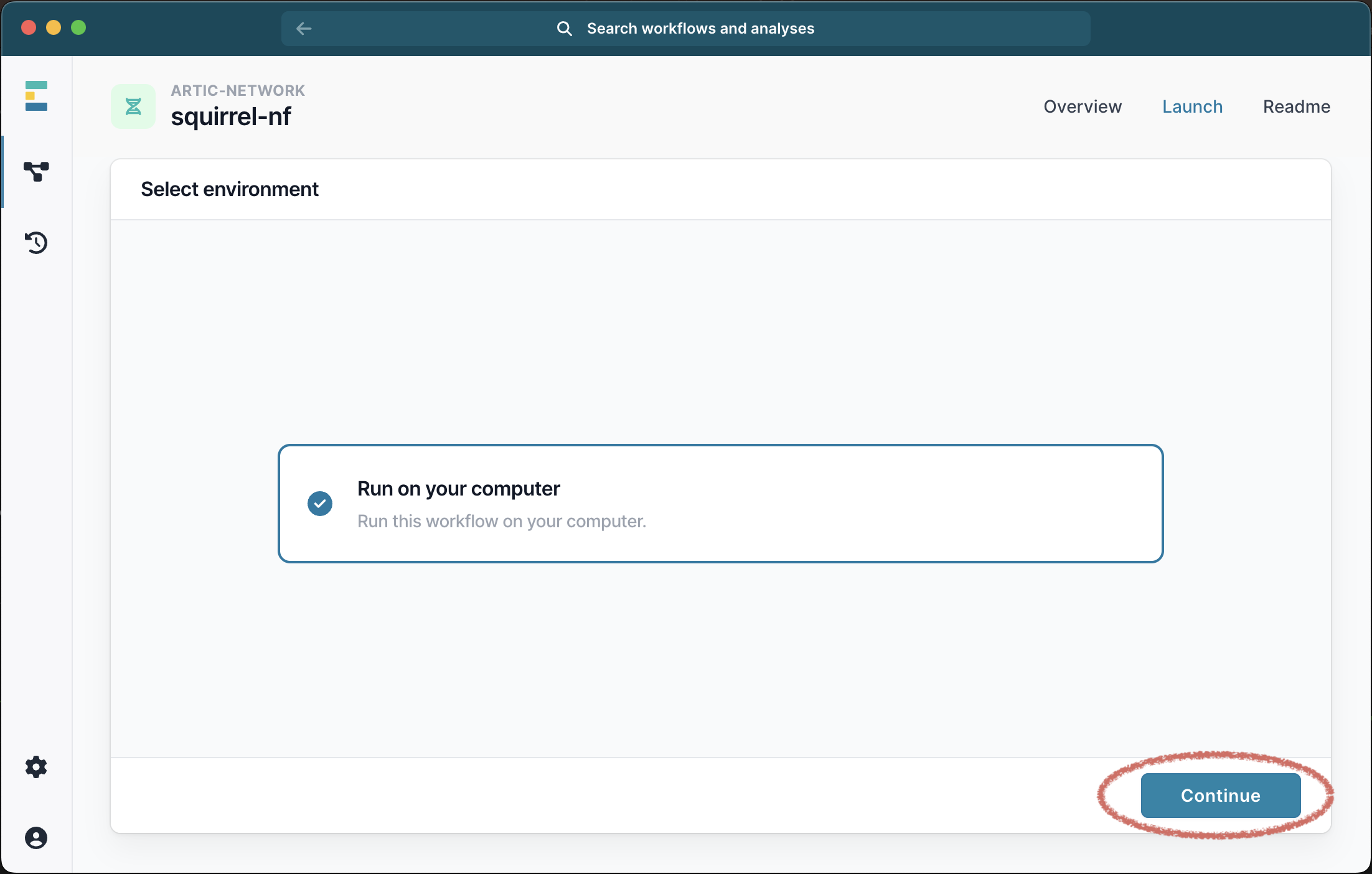
This will bring up a menu where you can provide the inputs for your analysis. The only required file is a single FASTA file containing all the sequences and outgroups for your analysis and you must also select the clade (i or ii) from the drop down list:
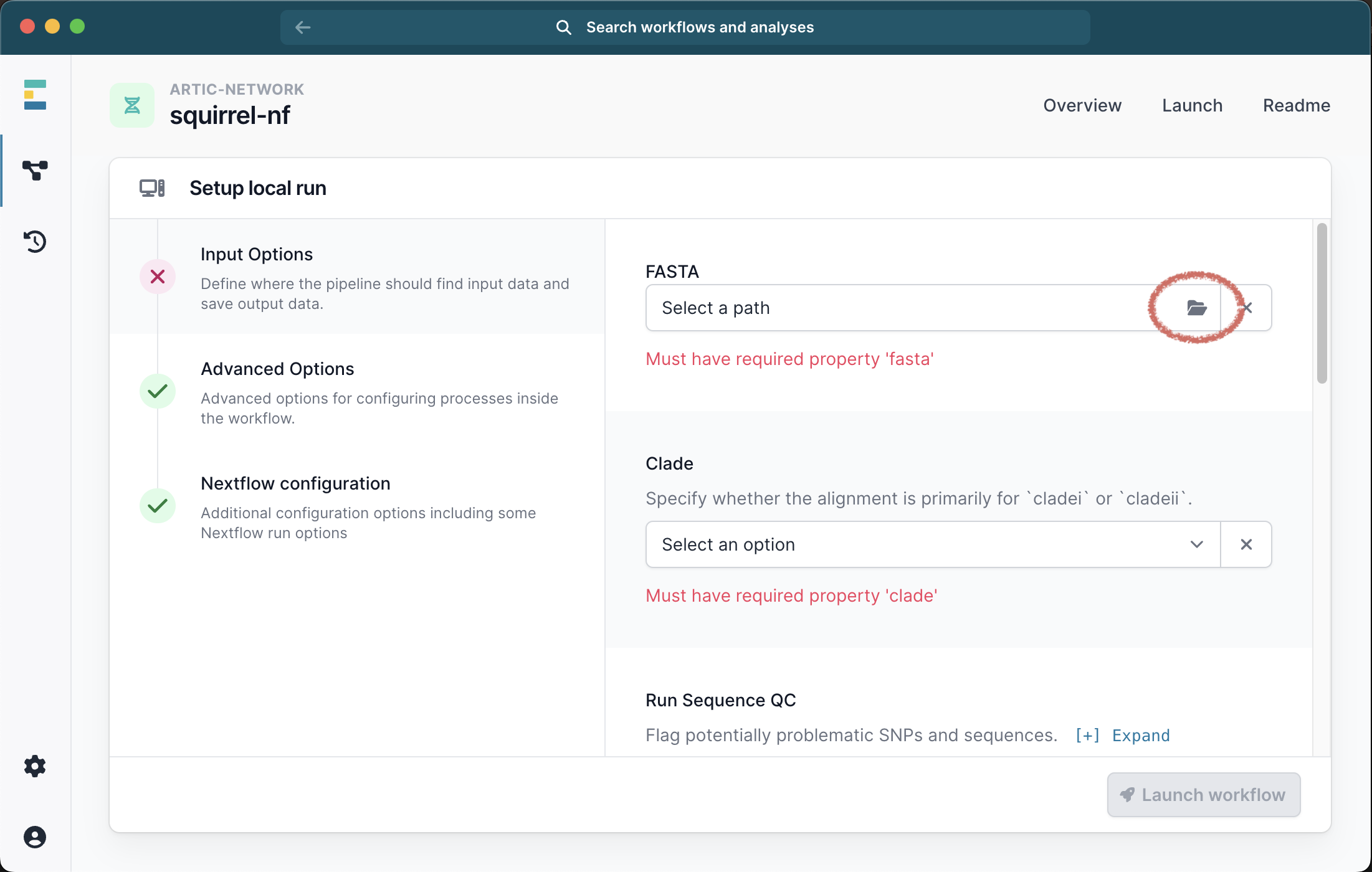
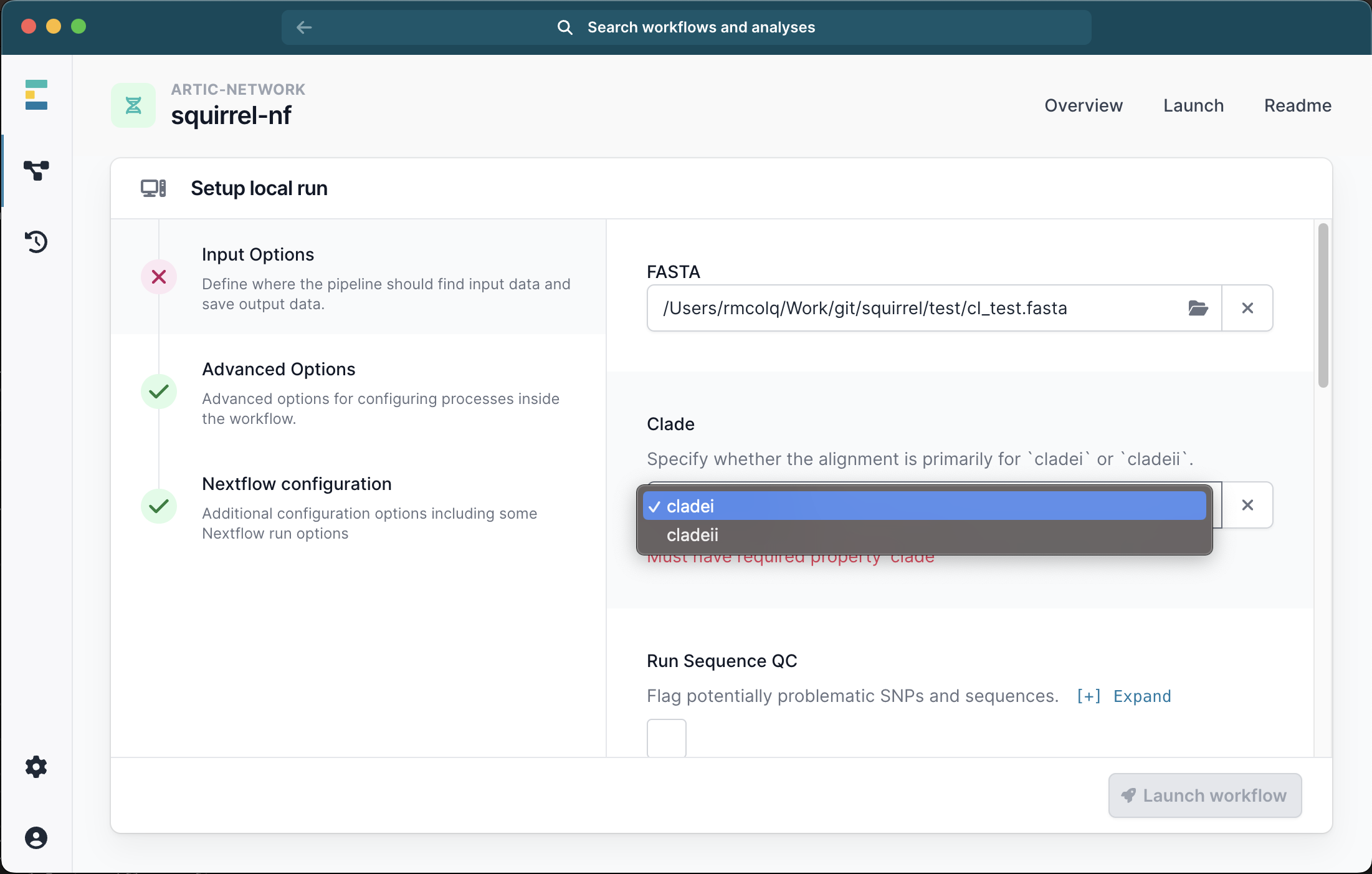
Running with just a FASTA file will generate an alignment of the input sequences. We recommend selecting the check box for Seq QC to check this alignment for problematic sites.
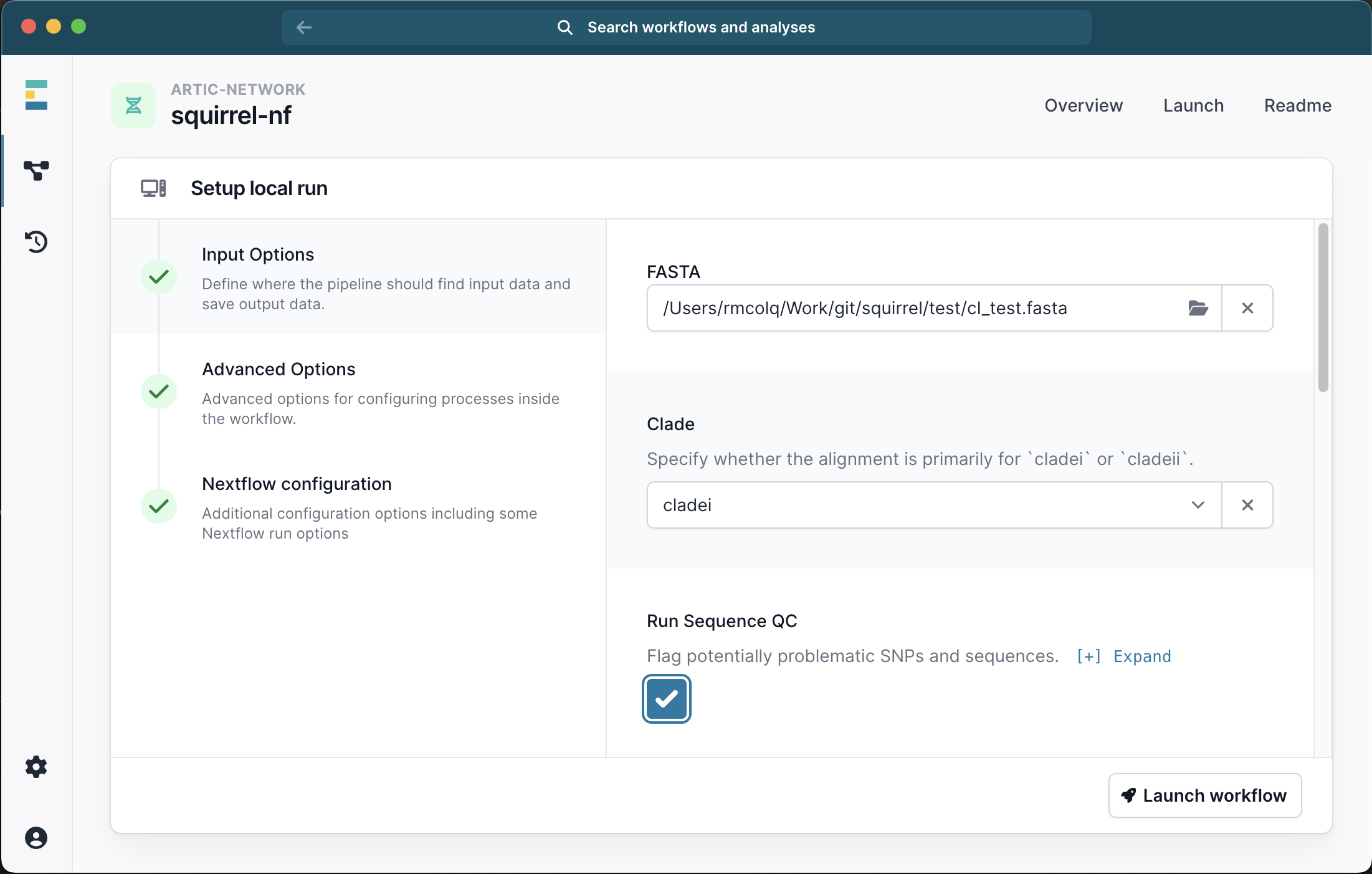
Scrolling down the menu, select the box to Run Phylo. At this point you have 2 options. EITHER you can select the check box to Include Background, in which case a default panel of clade-specific outgroups sequences will be used.
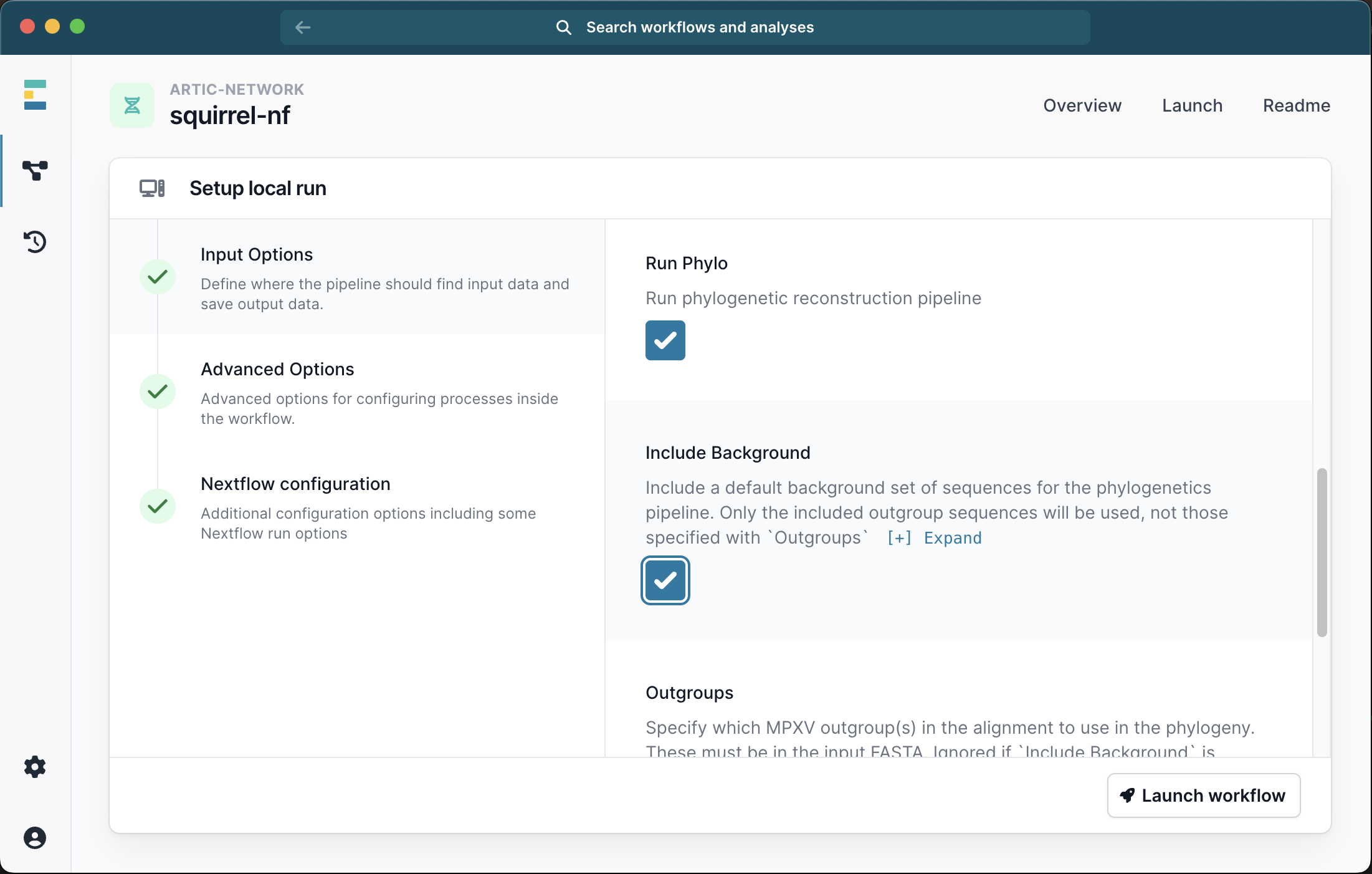
OR you can specify a number of outgroups IDs. These outgroups must be present in the FASTA file you provided and will be pruned out of the final alignment. For Clade I we recommend outgroups KJ642617,KJ642615,KJ642616 and for Clade IIb we recommend KJ642617,KJ642615. If you also selected the Include Background option your specified outgroups will be ignored.
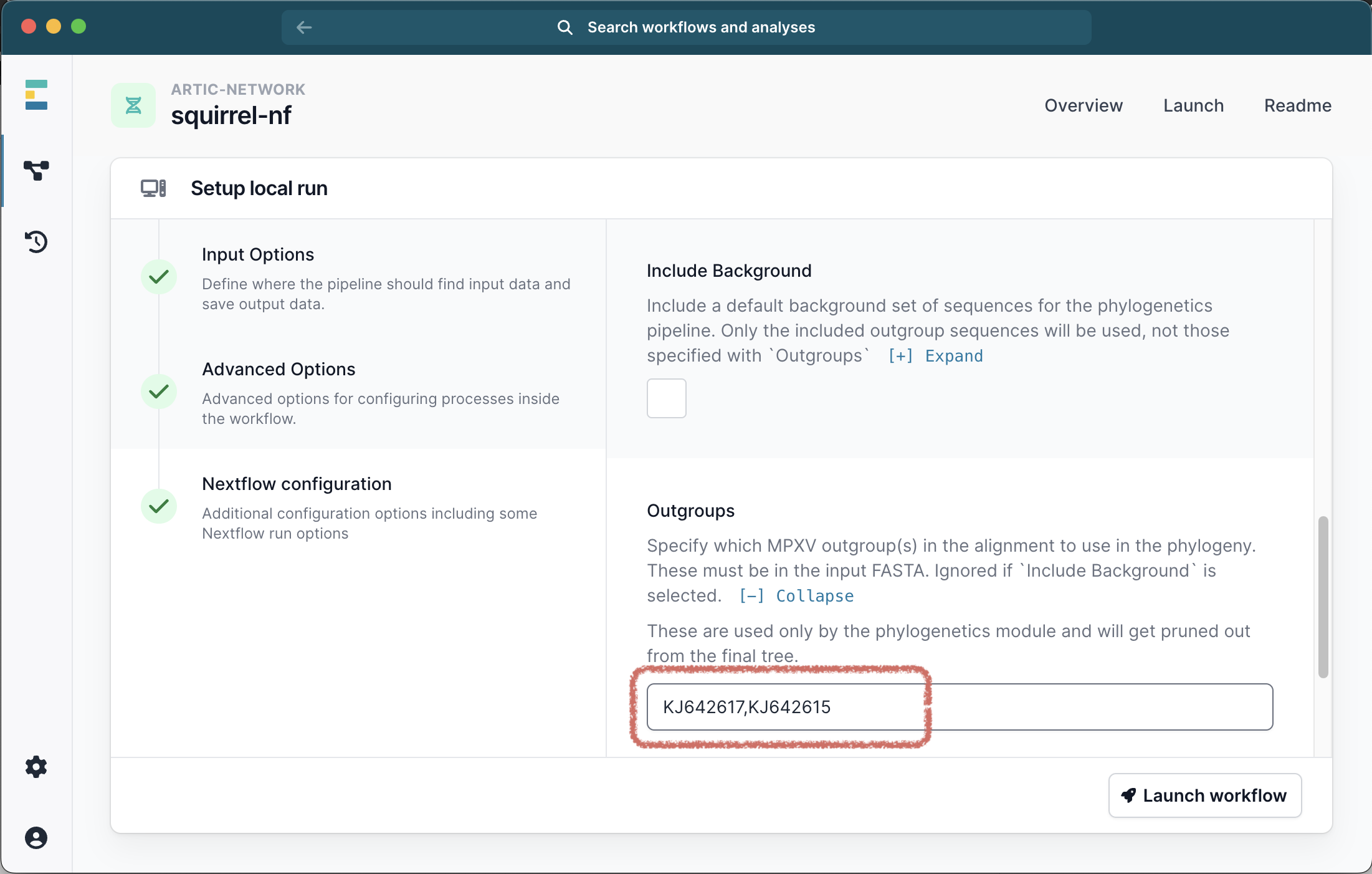
Optionally you can provide a different reference sequence, but this is usually unnecessary - a clade specific reference will be used by default. No Advanced Options or Nextflow Configuration options are required by default.
Click Launch Workflow and then Launch:
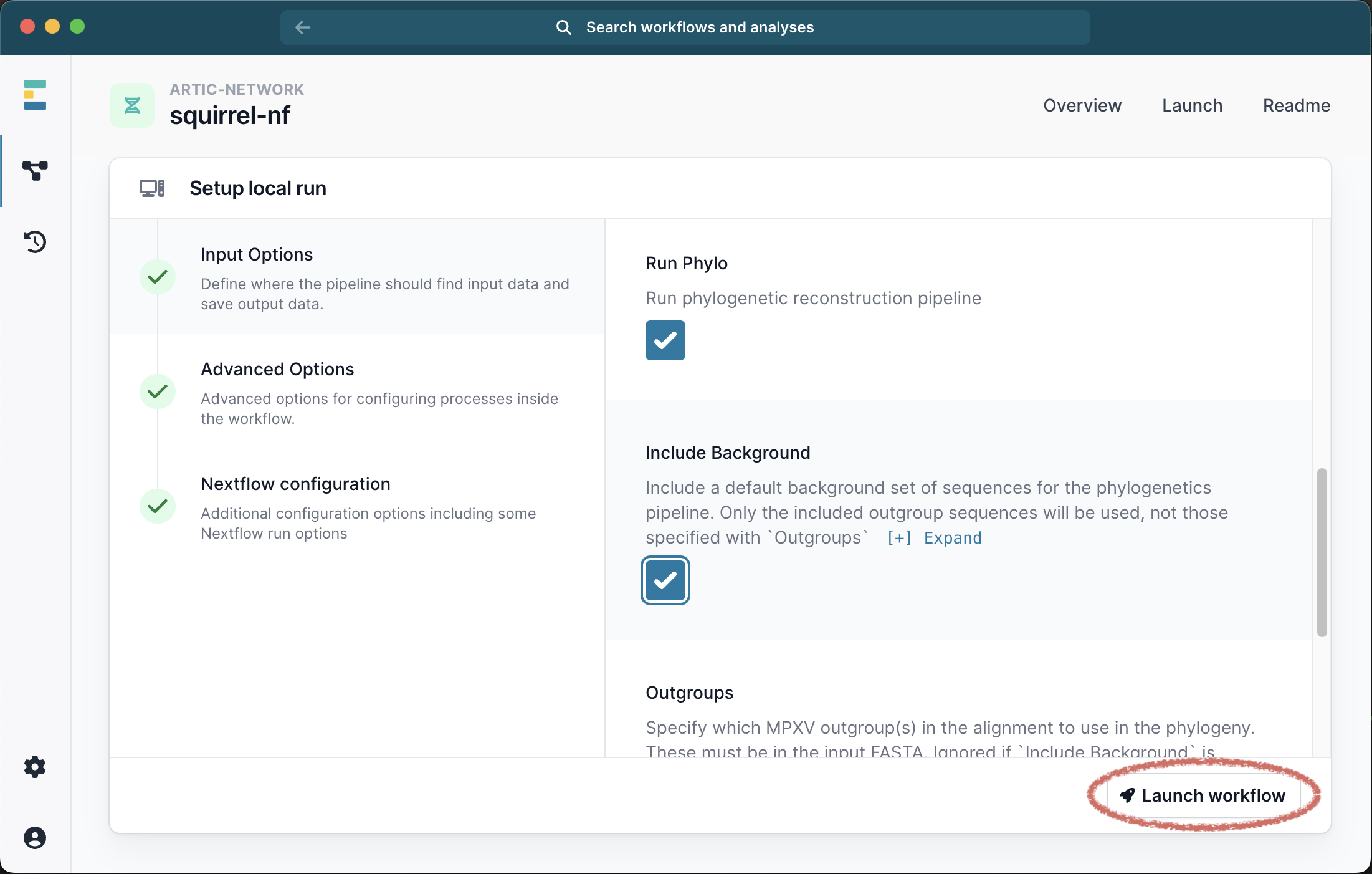
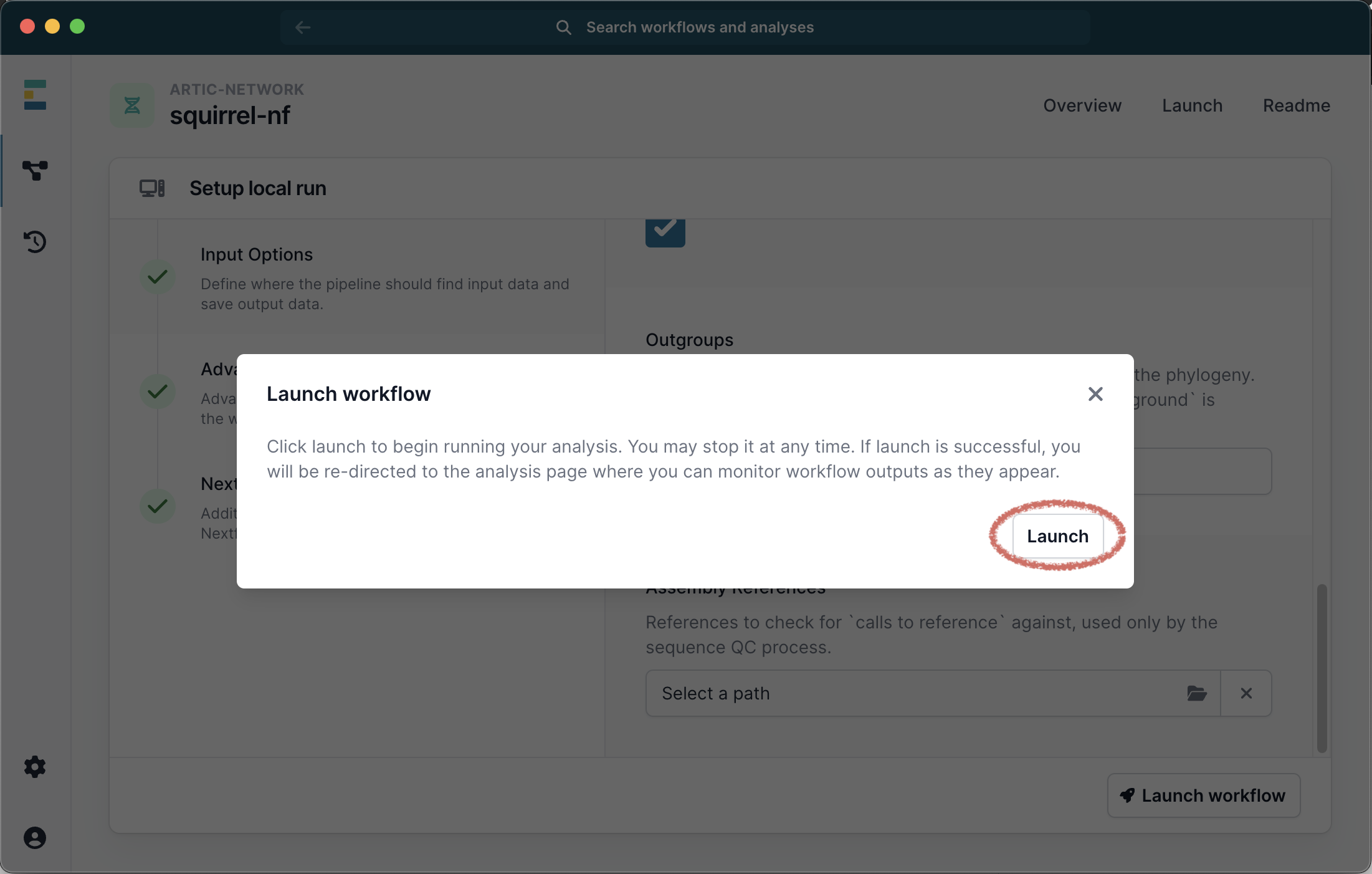
This will start the workflow. A progress bar is displayed with the run status but you will not be able to see the stdout that is generated on the command line.
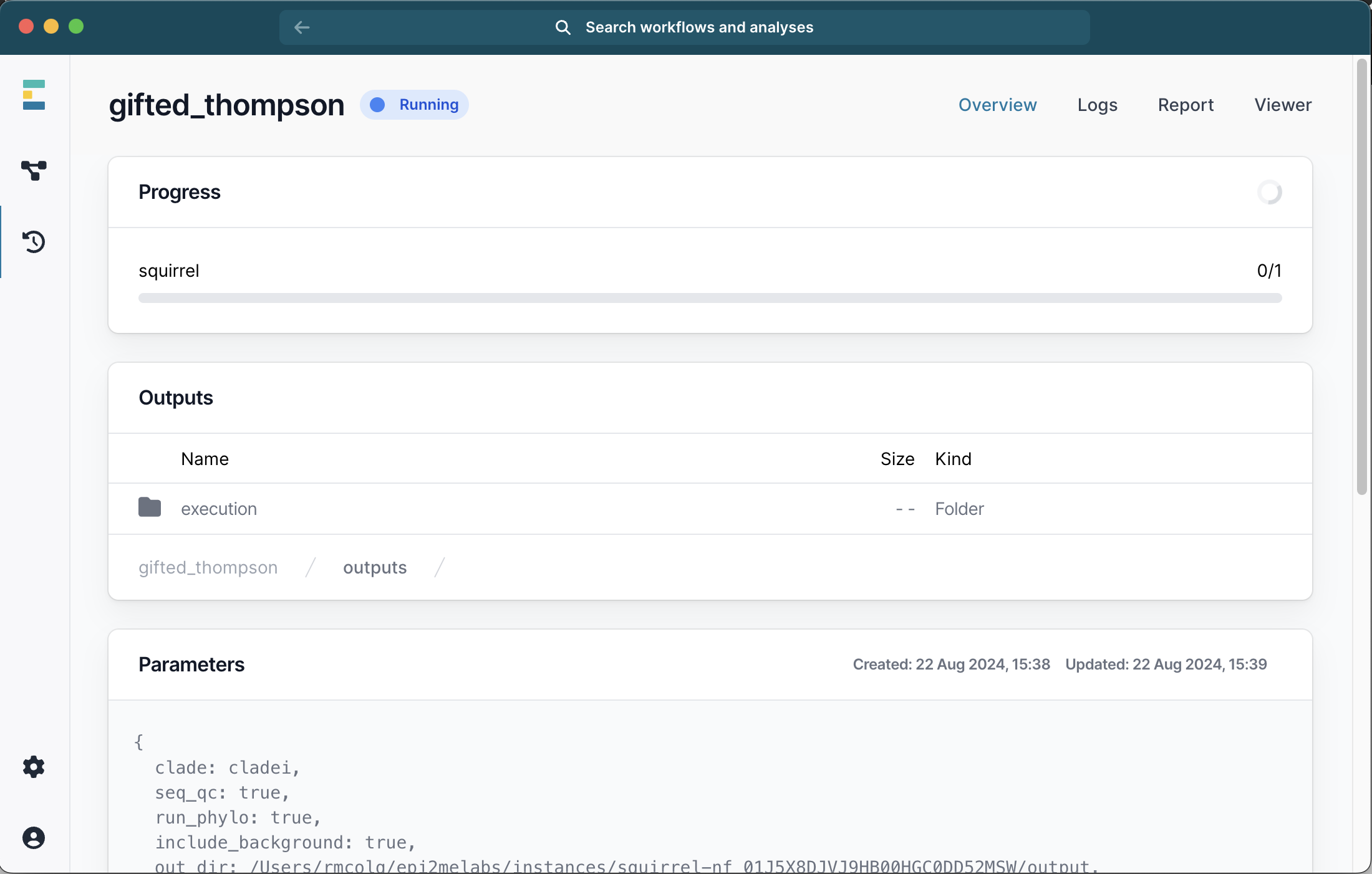
Once the run is completed, a number of files will be available and you can double-click to view them:
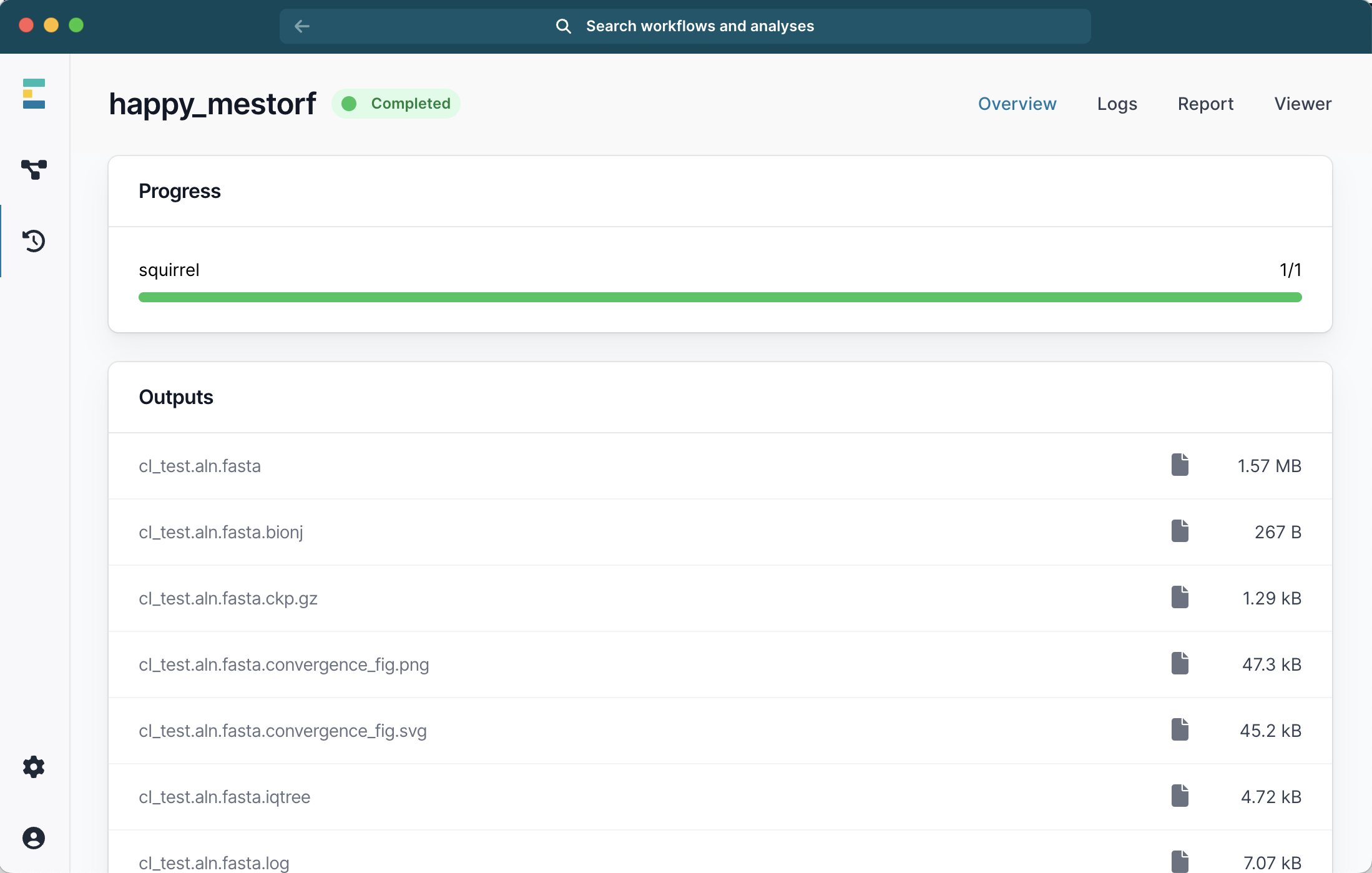
This includes a suggested_mask.csv file generated by the run with potentially problematic sites. If you start a new run with the same inputs and additionally provide this mask file in the menu, it will improve the alignment and phylogeny.
Pipeline description
Alignment
Squirrel maps each query genome in the input file against a reference genome specific to each clade using minimap2. Using gofasta, the mapping file is then converted into a multiple sequence alignment.
For Clade II, the reference used is NC_063383 and for Clade I, we use NC_003310, these are the RefSeq references for each of the clades on NCBI Genbank. This means that all coordinates within an alignment will be relative to these references. A benefit of this is that within a clade, alignment files and be combined without having to recalculate the alignment. Note however that insertions relative to the reference sequence will not be included in the alignment.
Masking
Before the final alignment is produced by squirrel, there are additional processing steps that include various masking options. By default, squirrel trims the alignment to 190788 at the end of the genome to mask out one of the inverted terminal repeat (ITR) regions and pads the end of the genome with N. This can be disabled with the no itr mask option.
Squirrel performs masking (replacement with N) on low-complexity or repetitive regions that have been characterised for Clade I and II. These regions are defined in to_mask.cladeii.csv and to_mask.cladei.csv respectively, and the relevant mask file will be selected depending on the clade specified. This masking is on by default but can be toggled off.
Squirrel can also accept an additional mask file in csv format (additional-mask), if there are flagged sites that you wish to mask within the alignment. These sites will be masked in conjuntion with the default masking files within squirrel. The format of the mask file fits with the features format from Geneious and at a minimum should contain the following fields: “Maximum”,”Minimum”.
QC mode
Squirrel can run quality control (QC) on the alignment and flag certain sites to the user that may need to be masked. We recommend that the user looks at these sites in an alignment viewer to judge whether the sites should be masked or not. If QC mode is toggled on, squirrel with check within the alignment for:
- Mutations that are adjacent to N bases The rationale for this is that N sites are usually a product of low coverage regions. Mutations that occur directly adjacent to low coverage regions may be a result of mis-alignment prior to the low coverage masking and may not be real SNPs.
- Unique mutations that clump together
If mutations are observed in only a single sequence in the genome, they are classed as unique mutations.
Usually mutations do not clump closely together and may suggest an alignment or assembly issue. If these mutations are not shared with any other sequences, they are flagged for masking.
Phylogenetic inference
Tree vizualisations are rendered using baltic
References
Heng Li, Minimap2: pairwise alignment for nucleotide sequences, Bioinformatics, Volume 34, Issue 18, September 2018, Pages 3094–3100, https://doi.org/10.1093/bioinformatics/bty191
Ben Jackson, gofasta: command-line utilities for genomic epidemiology research, Bioinformatics, Volume 38, Issue 16, August 2022, Pages 4033–4035, https://doi.org/10.1093/bioinformatics/btac424
Bui Quang Minh, Heiko A Schmidt, Olga Chernomor, Dominik Schrempf, Michael D Woodhams, Arndt von Haeseler, Robert Lanfear, IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era, Molecular Biology and Evolution, Volume 37, Issue 5, May 2020, Pages 1530–1534, https://doi.org/10.1093/molbev/msaa015
Gytis Dudas (2016) https://github.com/evogytis/baltic